Clear Sky Science · de
Ein absoluter Quantifizierungsatlas kleiner nichtkodierender RNAs in verschiedenen Säugetiergeweben und Zelllinien
Warum winzige RNA-Moleküle wichtig sind
In jeder Zelle entscheiden Flotten winziger RNA-Moleküle mit, welche Gene ein- oder ausgeschaltet werden. Diese kleinen nichtkodierenden RNAs wirken wie Dimmer für genetische Programme und prägen Entwicklung, Organfunktionen und Erkrankungen. Trotz leistungsfähiger Sequenzierungstechnologien fiel es Forschern bislang schwer, die exakte Anzahl dieser Moleküle in unterschiedlichen Zellen und Geweben zu bestimmen. Diese Studie stellt eine genauere Methode zur Zählung vor und erstellt einen detaillierten Atlas, der ihre tatsächliche Häufigkeit in vielen Säugetiergeweben und gängigen Laborzelllinien zeigt.
Eine klarere Methode zum Zählen kleiner RNAs
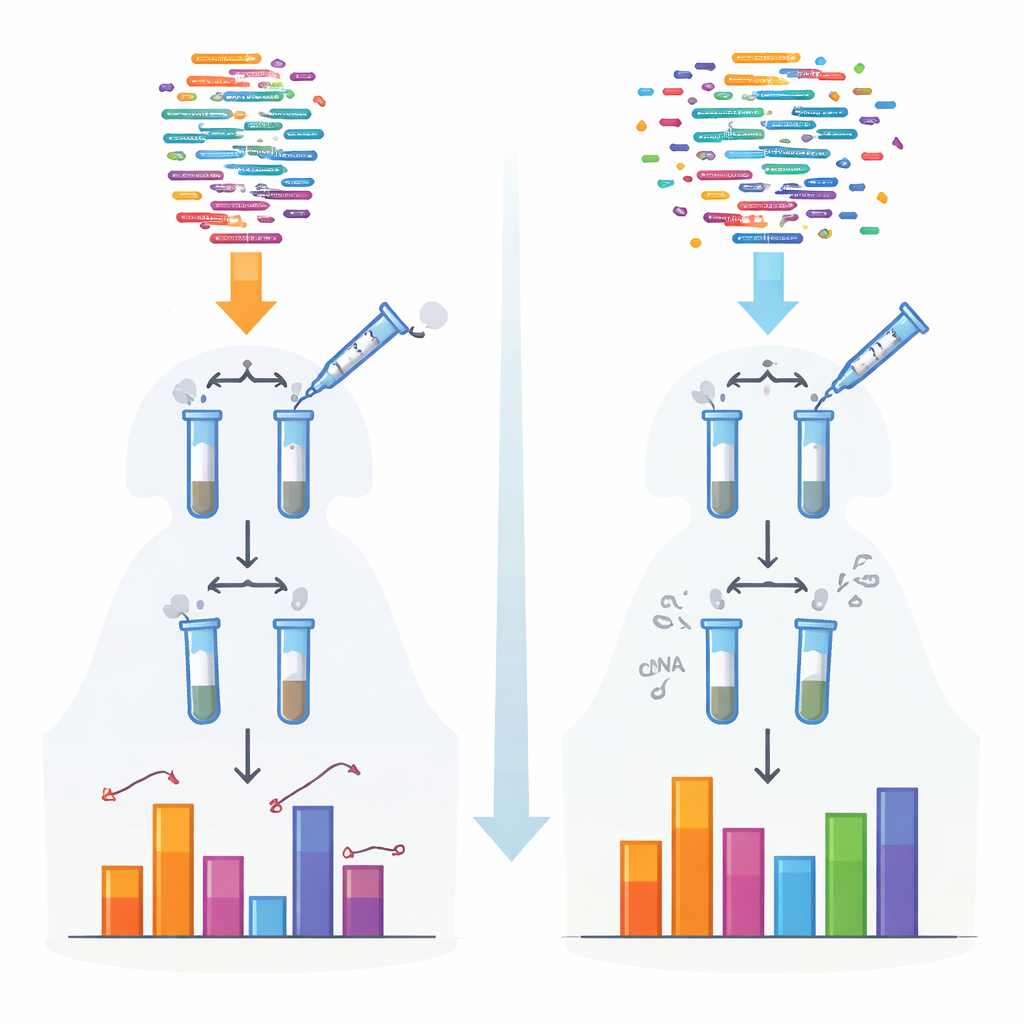
Traditionelle Methoden zur Sequenzierung kleiner RNAs beruhen auf Enzymen, die Adapterstücke anfügen, bevor die Moleküle gelesen werden. Diese Enzyme bevorzugen bestimmte Formen und chemische Enden, sodass einige RNAs effizient erfasst werden, während andere übersehen oder unterzählt werden. Dieser Bias trifft besonders bestimmte Klassen wie piRNAs und pflanzliche kleine RNAs, die schützende chemische Kappen an ihren Enden tragen. Die Autorinnen und Autoren entwickelten ein neues Protokoll namens 4NBoost, das die Adapter und Reaktionsbedingungen so überarbeitet, dass diese Präferenzen ausgeglichen werden, und integrierte molekulare Barcodes hinzufügt, um echte Moleküle von während der Amplifikation entstandenen Kopien zu unterscheiden.
Ein Protokoll in ein Messinstrument verwandeln
Um 4NBoost von einer relativen Messgröße in ein tatsächliches Messinstrument zu verwandeln, fügte das Team sorgfältig entworfene synthetische „Spike‑in“-RNAs in bekannten Konzentrationen über einen sehr weiten Bereich hinzu. Durch den Vergleich, wie oft jeder Spike‑in vom Sequenzierer gelesen wurde und wie viel ursprünglich zugegeben wurde, bauten sie Standardkurven, die Lesezahlen in absolute Molekülzahlen umrechnen. Tests mit unterschiedlichen Spike‑in-Mischungen und zusätzlichen Kontroll-RNAs zeigten, dass 4NBoost die Häufigkeit über mehrere Größenordnungen hinweg genau verfolgen kann, einschließlich RNAs mit problematischen chemischen Modifikationen. Selbst bei Startmengen von nur einem Nanogramm Gesamt‑RNA erfasste die Methode die Landschaft der kleinen RNAs zuverlässig.
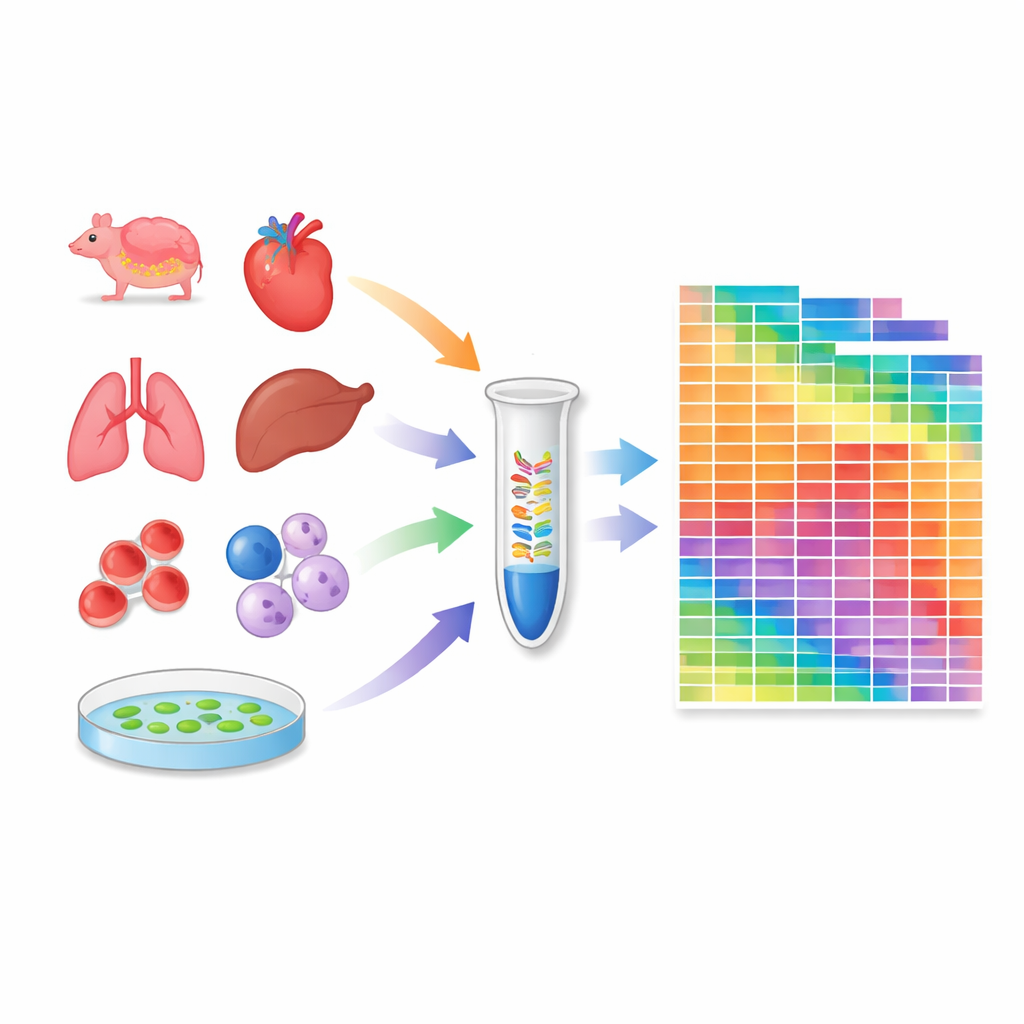
Einen Atlas über Gewebe und Zelllinien erstellen
Mit diesem kalibrierten Protokoll profilierte das Forschungsteam 259 Proben: 20 Gewebe von Mäusen, 18 von Krallenaffen, 24 häufig verwendete menschliche und Maus-Zelllinien sowie mehrere Gewebe der Modellpflanze Arabidopsis. Für jede Probe schätzten sie die absolute Molekülzahl für Tausende von microRNAs und piRNAs. Daraus ging hervor, wie viele verschiedene microRNA‑Arten in jedem Kontext vorhanden sind und wie sich ihre Gesamtmengen zwischen Geweben und Arten unterscheiden. Einige Zelllinien und Organe besitzen besonders reiche microRNA‑Repertoires, während andere, wie Blutzellen, von wenigen sehr häufigen Spezies dominiert werden. Der Atlas deckte zudem erhebliche Unterschiede zwischen Maus- und Affengeweben auf und unterstreicht, dass die Regulation durch kleine RNAs artspezifisch sein kann.
Alte Daten korrigieren und gängige Annahmen überarbeiten
Beim Vergleich des neuen Atlas mit populären kleinen‑RNA‑Datenbanken, die mit konventionellen Methoden erstellt wurden, traten auffällige Abweichungen zutage. Mehrere wichtige microRNA‑Familien — wie miR‑19 und miR‑29 — erwiesen sich als deutlich häufiger als bisher angenommen, während andere — etwa die viel untersuchten let‑7‑ und miR‑10‑Familien — oft überschätzt waren. Die Studie untersuchte außerdem, welcher „Arm“ jedes Vorläufer‑Haarnadelstruktur tatsächlich in Zellen verwendet wird, und fand Fälle, in denen aktuelle Annotationen die falsche Hauptstrang‑Variante listen. Um die Fülle bestehender verzerrter Datensätze zu retten, trainierten die Autorinnen und Autoren ein Modell des maschinellen Lernens, das lernt, wie konventionelle Messungen von 4NBoost abweichen, und diese dann mathematisch korrigiert, um besser die wahren Häufigkeiten widerzuspiegeln.
Eine öffentliche Ressource zum Erkunden kleiner RNAs
Alle 4NBoost‑Messungen und das Korrekturmodell stehen kostenlos über eine Online‑Plattform namens SmRNAQuant zur Verfügung. Forschende können absolute kleine‑RNA‑Werte für spezifische Gewebe, Zelllinien oder microRNAs durchsuchen oder herunterladen und eigene mit einem gängigen Kit aufbereitete Daten hochladen, um bias‑korrigierte Werte zu erhalten. Für Nicht‑Spezialisten lautet die zentrale Botschaft: Zählen ist wichtig — winzige Unterschiede in der Kopienzahl kleiner RNAs können den Unterschied zwischen aktiver Genregulation und keinem Effekt ausmachen. Indem sie verlässlichere Zahlen und einen Weg zur Korrektur älterer Daten bieten, legt diese Arbeit eine solidere quantitative Grundlage dafür, wie kleine RNAs normale Biologie und Krankheit prägen.
Zitation: Xiao, W., Zheng, Y., Zhang, H. et al. An absolute quantification atlas of small non-coding RNAs across diverse mammalian tissues and cell lines. Nat Commun 17, 2314 (2026). https://doi.org/10.1038/s41467-026-68812-7
Schlüsselwörter: microRNA-Quantifizierung, kleine nichtkodierende RNA, Bias bei RNA-Sequenzierung, Gewebe-Expressionsatlas, maschinelles Lernen Korrektur