Clear Sky Science · de
Strukturelle Grundlagen und pathologische Implikationen des dimeren OS9-SEL1L-HRD1 ERAD-Kernkomplexes
Die zelluläre Aufräumtruppe unter dem Mikroskop
In jeder unserer Zellen wandelt eine geschäftige Fabrik genetische Anweisungen in funktionsfähige Proteine um. Wie in jeder Fabrik passieren auch hier Fehler. Wenn Proteine falsch falten, können sie das System verstopfen und zu Krankheiten beitragen. Diese Studie richtet den Fokus auf eine der wichtigsten Qualitätskontrollmaschinen der Zelle – den SEL1L‑HRD1‑Komplex – um seine dreidimensionale Struktur im Detail zu zeigen und zu erklären, wie winzige genetische Veränderungen diese Maschine zerstören und möglicherweise zu menschlichen Erkrankungen führen können.
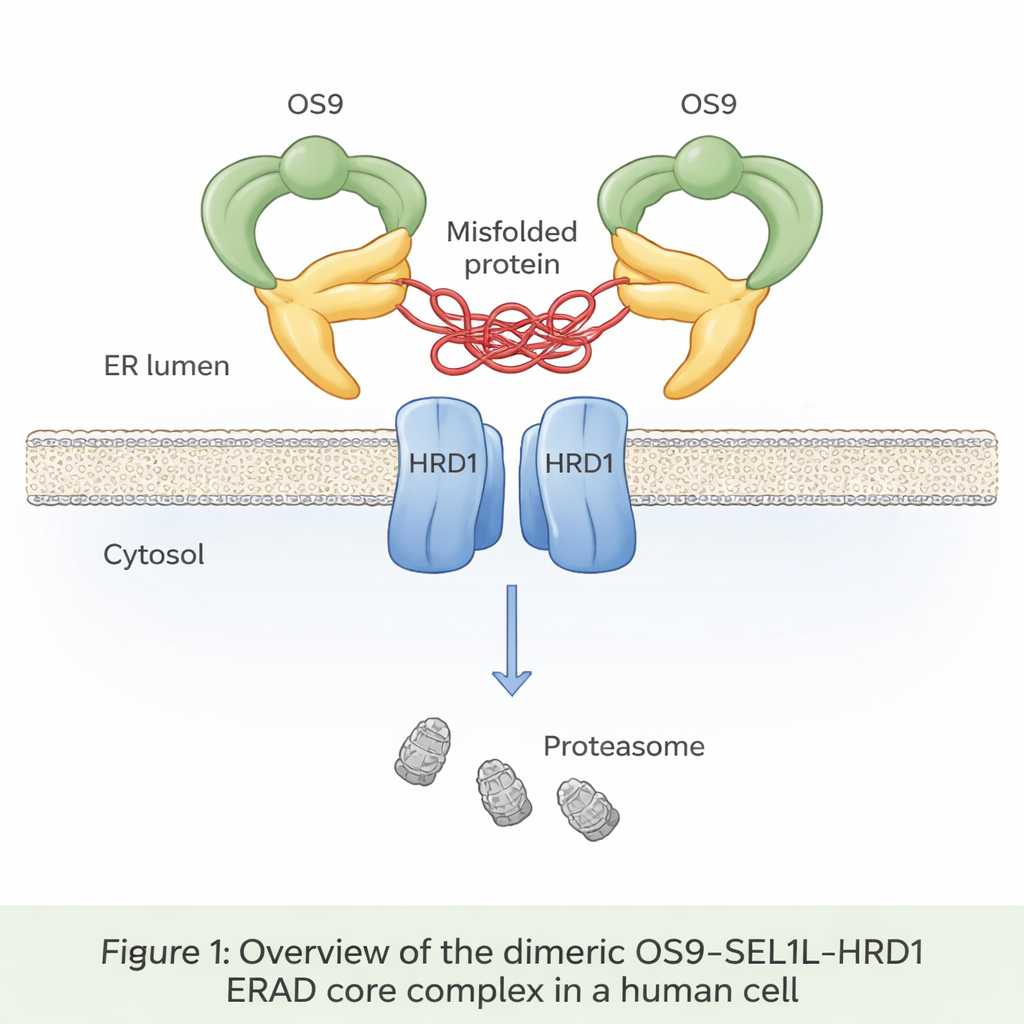
Ein verborgenes Förderband in der Zelle
Bis zu einem Drittel aller neu gebildeten Proteine gelangt in ein Kompartiment, das endoplasmatische Retikulum (ER) genannt wird, wo sie gefaltet und überprüft werden. Fehlgefaltete Proteine werden normalerweise erkannt, aus dem ER zurückgezogen und in einem Prozess abgebaut, der als ER‑assoziierter Abbau (ERAD) bekannt ist. Im Zentrum eines wichtigen ERAD‑Wegs stehen drei Proteine: OS9, SEL1L und HRD1. OS9 fungiert als Sensor für fehlerhafte, zuckermarkierte Proteine; SEL1L dient als Gerüst; und HRD1 markiert die ausgesonderten Proteine mit kleinen Ubiquitin‑Flaggen, die sie für die Zerstörung durch die zellulären Entsorgungseinheiten, die Proteasomen, kennzeichnen. Bis jetzt jedoch hatte noch niemand in atomarer Auflösung gesehen, wie diese drei Komponenten in menschlichen Zellen zusammenpassen.
Die Form der Kernmaschine enthüllen
Die Autoren nutzten Kryo‑Elektronenmikroskopie, eine Technik, die schockgefrostete Moleküle mit nahezu atomarer Auflösung abbildet, um den aus menschlichen Zellen gereinigten OS9‑SEL1L‑HRD1‑Komplex zu visualisieren. Sie entdeckten, dass er ein Dimer bildet – im Wesentlichen zwei identische Kopien, die miteinander verbunden sind – statt als Einzelteil zu bestehen. Auf der Lumen‑Seite des ER (der Innenseite des ER) setzen sich zwei OS9‑ und zwei SEL1L‑Moleküle zu einem krabbenklaueähnlichen Ring mit einer zentralen Öffnung zusammen, die offenbar dazu gedacht ist, fehlgefaltete Proteine zu erfassen. Innerhalb der Membran selbst paaren sich zwei HRD1‑Moleküle und bilden einen gemeinsamen Kanal. Diese Anordnung positioniert die „Klaue“ direkt über der HRD1‑Tür und schafft so einen durchgehenden Pfad, damit fehlgefaltete Proteine vom ER‑Lumen durch die Membran bis zur Zerstörung im Zytosol gelangen können.
Wie winzige Veränderungen ein großes System zerstören
Da Mutationen in SEL1L und HRD1 bei Patienten mit schweren neuroentwicklungsbedingten und anderen Störungen gefunden wurden, kartierte das Team mehrere krankheitsassoziierte Varianten auf ihrer Struktur und testete ihr Verhalten in Zellen. Zwei SEL1L‑Mutationen, G585D und S658P, liegen direkt an den Kontaktstellen zu OS9 bzw. HRD1. In Zellversuchen hob G585D nahezu SEL1Ls Fähigkeit auf, an OS9 zu binden, während S658P seine Bindung an HRD1 stark schwächte; die Kombination beider Mutationen zerstörte den Kernkomplex im Wesentlichen, ließ jedoch andere Partner unbeeinträchtigt. Infolgedessen hatten die Zellen Schwierigkeiten, ein fehlgefaltetes Hormonvorläuferprotein zu markieren und zu entfernen, sodass fehlerhafte Proteine bestehen blieben.
Eine Krankheitsmutation im Membrankanälchen
Die Struktur zeigt außerdem, dass das transmembrane Segment 3 von HRD1 die Schlüsseloberfläche ist, an der sich zwei HRD1‑Moleküle zur Bildung des Kanals begegnen. Die Forscher bauten an spezifischen Positionen Cystein‑„Griffe“ ein und nutzten chemische Quervernetzung, um zu bestätigen, dass diese Regionen in lebenden Zellen in engen Kontakt treten, was beweist, dass HRD1 in vivo tatsächlich dimerisiert. Als sie eine einzelne, hoch konservierte Aminosäure (T93) an dieser Schnittstelle störten, fiel das Dimer auseinander und die ERAD‑Aktivität brach zusammen, obwohl der Komplex weiterhin mit OS9 und SEL1L assembliert war. Anschließend untersuchten sie eine neu entdeckte Patientenvariante, HRD1 A91D, die bei einem Kind mit Herz‑ und Lungenproblemen gefunden wurde. Diese Veränderung, ebenfalls in der Dimer‑Schnittstelle, reduzierte die HRD1‑Dimerisierung um etwa die Hälfte und beeinträchtigte die Beseitigung fehlgefalteter Proteine stark, wiederum ohne die generelle Partnerbindung zu stören.
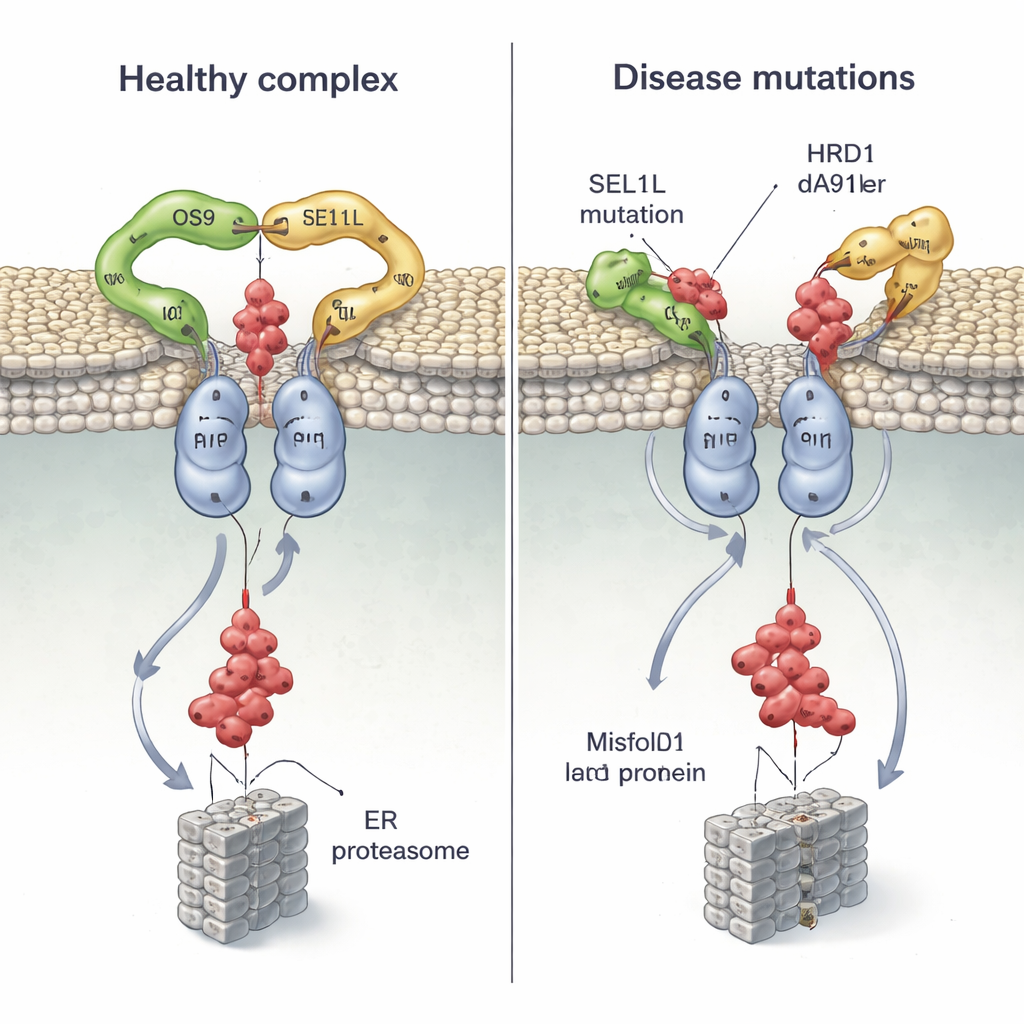
Ein neuer Blick auf Protein‑Qualitätskontrolle und Krankheit
Durch die Kombination struktureller Biologie mit zellbasierten Tests zeigt diese Arbeit, dass der OS9‑SEL1L‑HRD1‑Komplex als gepaarte, dimerische Maschine arbeitet: eine klaueähnliche Auffangvorrichtung, die an einen gemeinsamen Kanal gebunden ist und fehlerhafte Proteine aus dem ER heraustransportiert. Mutationen, die den Griff der Klaue lockern oder das HRD1‑Paar destabilisieren, verändern nicht nur die Effizienz – sie können das System effektiv blockieren, sodass beschädigte Proteine sich anreichern und zur menschlichen Krankheit beitragen. Für Nicht‑Spezialisten lautet die Kernbotschaft, dass schon einzelne Buchstabenänderungen in unserer DNA die Form essentieller zellulärer Maschinen subtil verformen können, mit weitreichenden Folgen für Gehirnentwicklung, Immunfunktion und Organleistung.
Zitation: Lin, L.L., Maldosevic, E., Zhou, L.E. et al. Structural basis and pathological implications of the dimeric OS9-SEL1L-HRD1 ERAD Core Complex. Nat Commun 17, 2064 (2026). https://doi.org/10.1038/s41467-026-68777-7
Schlüsselwörter: Protein‑Qualitätskontrolle, Endoplasmatisches Retikulum, ERAD, SEL1L-HRD1-Komplex, Proteinfehlfaltung