Clear Sky Science · de
Zuverlässige Vorhersage von Enzym-Kommissionsnummern mithilfe eines hierarchischen interpretierbaren Transformers
Warum die Vorhersage von Enzymaufgaben wichtig ist
Jede lebende Zelle funktioniert dank zahlloser winziger chemischer Maschinen, den Enzymen. Jedes Enzym hat eine spezifische „Aufgabe“, die in einer Enzyme Commission (EC)-Nummer codiert ist — ein vierteiliger Code, ein wenig wie eine Postanschrift. Die korrekte Zuordnung von EC-Nummern ist entscheidend, um Stoffwechselwege zu verstehen, neue Medikamente zu entwerfen, Mikroben so zu konstruieren, dass sie Treibstoffe oder Kunststoffalternativen herstellen, und um nachzuvollziehen, wie Ökosysteme Chemikalien umsetzen. Experimentelle Bestimmungen von Enzymfunktionen sind jedoch langsam und teuer. Diese Studie stellt HIT-EC vor, ein neues KI-Modell, das EC-Nummern aus Proteinsequenzen zuverlässig vorhersagen kann und gleichzeitig erklärt, warum es jede einzelne Vorhersage getroffen hat.
Ein Postleitzahl-System für Enzymaufgaben
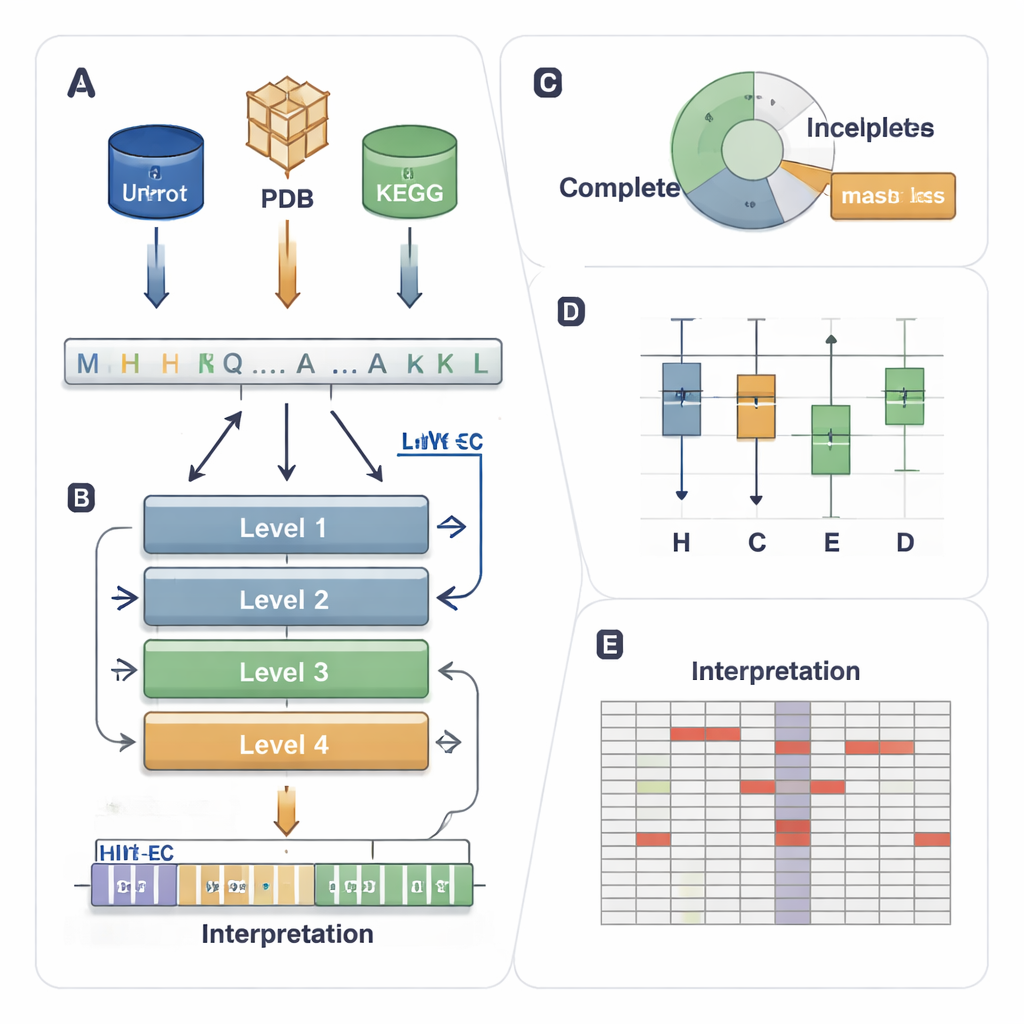
Das EC-System weist jedem Enzym einen vierstufigen Code wie 1.1.1.37 zu. Die erste Ziffer gibt eine grobe Klasse an (zum Beispiel Enzyme, die Elektronen bewegen oder Gruppen übertragen), die folgenden Ziffern beschreiben immer präzisere Reaktionsmerkmale. Diese Hierarchie ist mächtig, stellt aber auch eine anspruchsvolle Vorhersageaufgabe: Ein Modell muss alle vier Ebenen korrekt treffen für tausende mögliche Codes, selbst wenn manche Enzyme selten sind oder Datenbanken nur teilweise annotierte Einträge enthalten (zum Beispiel 3.5.-.-, wenn die detaillierten Ebenen fehlen). Bestehende Computermethoden verwenden entweder 3D-Struktur, Sequenzähnlichkeit oder Deep Learning, haben jedoch oft Probleme mit seltenen Enzymen, ignorieren teilweise annotierte Daten und verhalten sich meist wie „Black Boxes“, die kaum Einblick geben, warum sie eine Entscheidung treffen.
Ein vierstöckiges KI-Modell, das der EC-Leiter folgt
HIT-EC (Hierarchical Interpretable Transformer for EC prediction) ist so konstruiert, dass es die vierstufige EC-Hierarchie widerspiegelt. Es nimmt eine rohe Proteinsequenz und leitet sie durch vier Transformer-Schichten, von denen jede auf eine EC-Ebene fokussiert ist. Lokale Verbindungen koppeln jede Ebene an die vorhergehende, sodass eine feinere Entscheidung (die vierte Ziffer) mit breiteren Entscheidungen (erste und zweite Ziffer) konsistent sein muss. Parallel dazu erhält ein globaler Fluss den vollständigen Sequenzkontext in jedem Schritt. Das Modell kann auch mit Sequenzen trainiert werden, die unvollständige Labels haben, indem ein „masked loss“ verwendet wird, der fehlende EC-Ebenen einfach ignoriert, anstatt die Sequenz zu verwerfen. So kann HIT-EC aus dem großen Anteil an Proteinen in kuratierten Datenbanken lernen, die nur teilweise annotiert sind.
Führend bei Genauigkeit und Geschwindigkeit
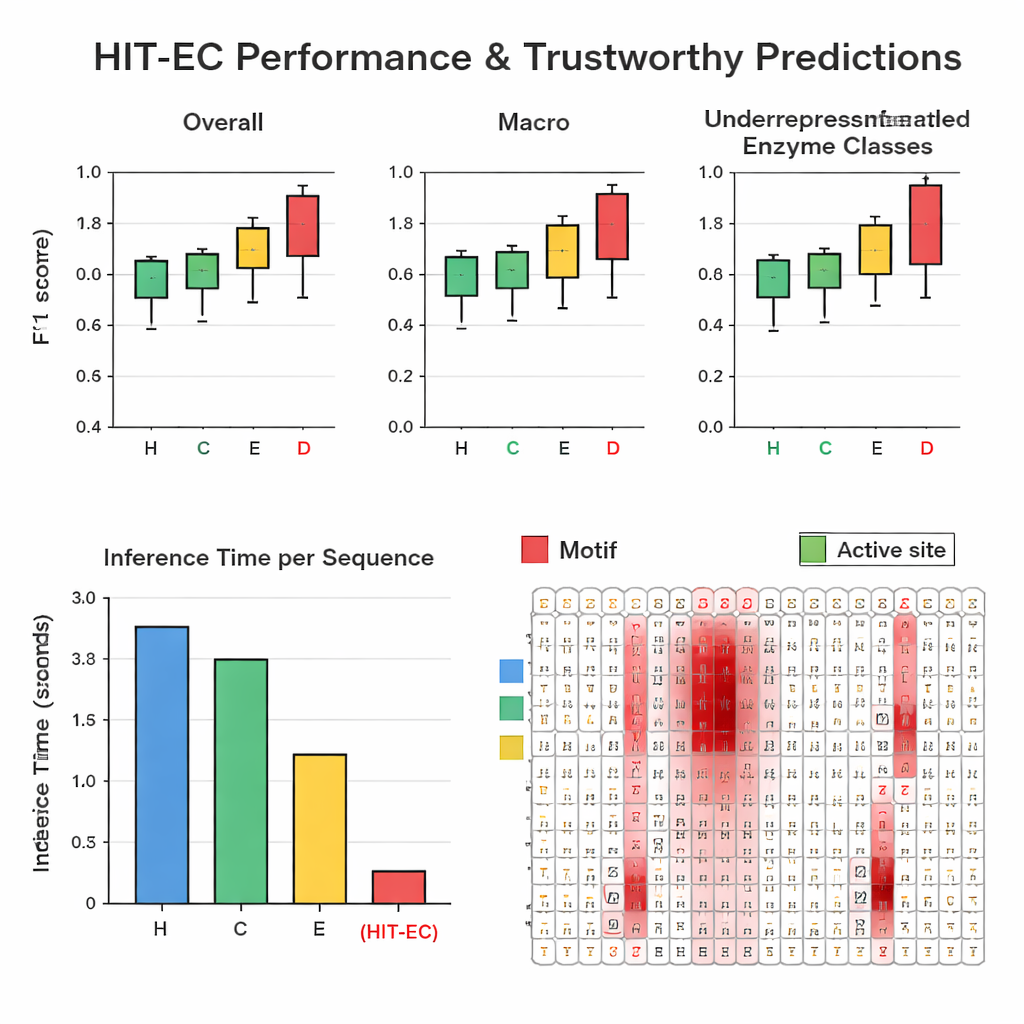
Die Autoren stellten ein großes, sorgfältig gefiltertes Datenset von rund 200.000 Enzymen mit 1.938 verschiedenen EC-Nummern aus Swiss-Prot und dem Protein Data Bank zusammen. In wiederholten Hold-out-Tests übertraf HIT-EC drei führende Methoden (CLEAN, ECPICK und DeepECtransformer) sowohl bei den Gesamt- als auch bei den klassenweisen F1-Scores, die das Gleichgewicht zwischen Treffern und Fehlalarmen messen. Besonders stark war das Modell bei unterrepräsentierten EC-Codes mit 25 oder weniger bekannten Beispielen, bei denen frühere Methoden oft versagen. HIT-EC generalisierte außerdem gut auf neue Enzyme, die nach dem Training zu Swiss-Prot hinzugefügt wurden, und auf vollständige Genome verschiedener Bakterien, darunter gut untersuchte Stämme von Escherichia coli, Bacillus subtilis und Mycobacterium tuberculosis. Trotz seiner Komplexität war das Modell sehr effizient: Auf einer Standard-GPU verarbeitete es ein Protein in etwa 38 Millisekunden — um Größenordnungen schneller als einige Konkurrenten, die auf langsamere Ähnlichkeitssuchen oder Ensembles vieler Modelle angewiesen sind.
Zeigen, worauf das Modell „sieht“
Um seine Vorhersagen vertrauenswürdig zu machen, ist HIT-EC so ausgelegt, dass es anzeigt, welche Aminosäuren in der Sequenz jede EC-Ebenen-Entscheidung beeinflusst haben. Die Autoren entwickelten einen Interpretationspfad, der Aufmerksamkeitsgewichte mit Gradienteninformationen kombiniert, um für jede Position eine Wichtigkeitsskala zu berechnen. Sie validierten diese Scores an gut charakterisierten Enzymfamilien. Beispielsweise hob HIT-EC in einer Cytochrom-P450-Familie (CYP106A2) bekannte funktionelle Motive hervor, wie Bereiche für Sauerstoffbindung und Häm-Bindung, und identifizierte ein subtiles EXXR-Motiv, das ein Vergleichsmodell verpasst hatte. Für klassische Vertreter jeder obersten EC-Klasse — etwa Alkohol-Dehydrogenase, Hexokinase und Carboanhydrase — leuchteten die Relevanzscores an Lehrbuch-signaturmotiven und Substratbindungsstellen auf. Diese Interpretationen liefern biochemische „Belege“, dass das Modell seine Entscheidungen auf sinnvolle Merkmale stützt und nicht auf zufällige Korrelationen.
Leitfaden für die Arbeit an seltenen und neu auftretenden Enzymen
Das Team testete HIT-EC zusätzlich an zwei wenig untersuchten Enzymen, die für die Umweltsanierung wichtig sind: einem Cytochrom P450, das am Abbau aromatischer Schadstoffe beteiligt ist, und einer PET-abbauenden Hydrolase aus Streptomyces, die bei der Zersetzung plastikbezogener Moleküle hilft. Beide Enzyme waren experimentell charakterisiert worden, besaßen aber keine offiziellen EC-Zuweisungen. HIT-EC sagte die erwarteten EC-Nummern korrekt voraus und hob Motivmuster sowie katalytische Reste hervor, die mit strukturellen und biochemischen Erkenntnissen übereinstimmen. Insgesamt zeigt die Arbeit, dass HIT-EC nicht nur EC-Nummern genauer und schneller zuweisen kann als aktuelle Werkzeuge — insbesondere für seltene Funktionen —, sondern auch aufzeigt, warum ein bestimmtes Enzym eine gegebene chemische Aufgabe ausführt. Diese Kombination aus Leistung und Interpretierbarkeit macht es zu einem vielversprechenden Instrument für großflächige, verlässliche Enzymannotation in Genomik, Biotechnologie und Umweltforschung.
Zitation: Dumontet, L., Han, SR., Lee, J.H. et al. Trustworthy prediction of enzyme commission numbers using a hierarchical interpretable transformer. Nat Commun 17, 1146 (2026). https://doi.org/10.1038/s41467-026-68727-3
Schlüsselwörter: Vorhersage von Enzymfunktionen, Deep Learning in der Biologie, Transformer-Modelle, Proteinannotation, Enzyme für Bioremediation