Clear Sky Science · de
Eine alternative EGFR-Aktivierung durch patientenabgeleitete R252C-Mutation fördert das Fortschreiten von Krebs
Wenn Zellantennen aus der Reihe tanzen
Warum wachsen manche Tumoren weiter, obwohl Patientinnen und Patienten bereits Chemotherapie und moderne Immuntherapien erhalten haben? Diese Studie begleitet einen Patienten mit Tumoren in Lunge und Gehirn und führt die Erkrankung auf eine winzige Veränderung in einer wichtigen zelloberflächenständigen "Antenne" namens EGFR zurück. Indem die Forschenden aufdecken, wie genau diese einzelne Mutation Wachstumssignale umkonfiguriert, erklären sie nicht nur die Aggressivität des Tumors, sondern zeigen auch, wie ein bereits zugelassenes Medikament, Afatinib, dieses Wachstum eindämmen kann.
Eine seltene Mutation mit großen Folgen
EGFR ist ein Rezeptor, der die Zellmembran durchspannt und Zellen hilft, auf Wachstumsreize zu reagieren. Viele Lungen- und Hirntumoren tragen Veränderungen im EGFR, doch die bekannten Mutationen liegen meist im Inneren des Proteins, in dem Teil, der wie ein enzymatischer Schalter funktioniert. Hier entdeckte das Team eine ungewöhnliche Veränderung an der Außenseite von EGFR, in dem Bereich, der normalerweise Wachstumsfaktoren bindet. Bei einem Patienten mit Lungenkrebs und Gliom war an Position 252 eine Aminosäure ausgetauscht worden: Arginin gegen Cystein — bezeichnet als EGFR R252C. Die Auswertung von Krebsdatenbanken zeigte diese Mutation bei einem kleinen Bruchteil der Gliom‑Patienten und nahezu nie in Lungenproben, was darauf hindeutet, dass sie selten, aber echt ist. Mithilfe von Geneditierung rekreierten die Autorinnen und Autoren diese exakte Mutation in mehreren menschlichen Hirn‑ und Lungenkrebszelllinien, um ihre Wirkung zu prüfen.
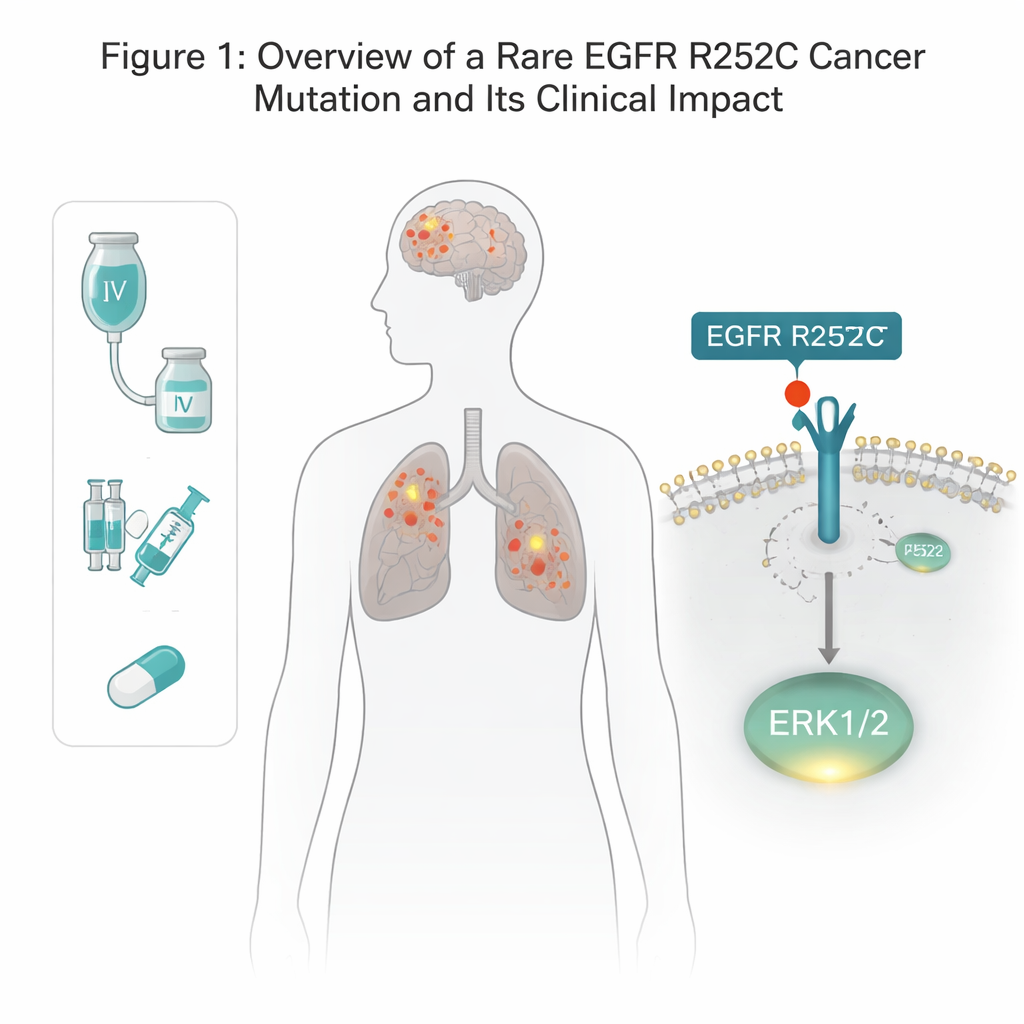
Eine neue Abkürzung zu Wachstumssignalen
Normalerweise muss EGFR mit einer zweiten Kopie dimerisieren und dann seinen eigenen inneren Schwanz mit Phosphatgruppen markieren, bevor es nachgeschaltete Wachstumspfade aktiviert. Überraschenderweise zeigte die R252C‑Variante dieses übliche Autophosphorylierungs‑Muster nicht. Stattdessen aktivierten Zellen mit EGFR R252C einen bestimmten Wachstumsregler, ERK1/2, deutlich stärker als normal, während andere klassische EGFR‑Wege — etwa AKT und STAT3 — weitgehend unverändert blieben. Die Blockade von ERK1/2 mit einem spezifischen Inhibitor beseitigte den zusätzlichen Wachstumsvorteil der R252C‑Zellen und bewies damit, dass ERK1/2 der Hauptmotor der tumorfördernden Wirkung dieser Mutation ist.
Den Rezeptor in einer Dauer‑Aktivform festklemmen
Um zu verstehen, wie eine äußere Veränderung eine so selektive Überaktivierung auslösen kann, kombinierten die Forschenden biochemische Tests mit Computersimulationen. Der Austausch zu Cystein an Position 252 bringt ein neues Cystein an die Außenseite von EGFR. Zwei solche Mutanten können eine Disulfidbrücke — eine Art molekularen Klammer — zwischen ihren C252‑Resten bilden und so stabil miteinander verbunden werden. Strukturelle Modellierungen zeigten, dass diese Bindung die Außenseite des Rezeptors in eine "V‑förmige", gestaffelte Anordnung zwingt, die der aktiven, ligandgebundenen Form sehr ähnlich ist, selbst ohne Wachstumsfaktor. Diese Ausrichtung setzt sich durch die membrandurchspannenden und unmittelbar intrazellulären Segmente fort und verdreht die internen Enzymdomänen in eine ungewöhnliche Konfiguration: Die aktiven Stellen zeigen zwar in die Zelle, sind aber zu weit voneinander entfernt, um sich effizient gegenseitig zu phosphorylieren. Stattdessen entsteht dadurch eine starke Andockfläche für ERK1/2, die es EGFR R252C erlaubt, ERK1/2 direkt zu phosphorylieren und so die übliche RAS–RAF–MEK‑Kaskade zu umgehen.
Von Mausmodellen bis zu einem einzelnen Patienten
Die Autorinnen und Autoren zeigten, dass Hirn‑ und Lungenkrebszellen mit EGFR R252C in Kultur schneller wuchsen und nach Implantation in Mäuse größere, aggressivere Tumoren bildeten als Zellen mit normalem EGFR. Sie testeten mehrere Generationen von EGFR‑Blockern. Nur Afatinib, ein Inhibitor der zweiten Generation, schaltete konsistent die ERK1/2‑Aktivierung aus und reduzierte das Tumorwachstum deutlich. In Mausmodellen von R252C‑getriebenen Hirn‑ und Lungentumoren verlangsamte Afatinib das Tumorwachstum und verlängerte das Überleben. Entscheidend ist, dass der ursprüngliche Patient — dessen Erkrankung trotz Chemotherapie, eines gefäßzielenden Wirkstoffs und Immuntherapie weiter fortgeschritten war — nach Umstellung auf Afatinib sowohl in Lunge als auch im Gehirn einen deutlichen Rückgang der Tumorlast zeigte und mehrere Jahre ohne Krankheitsprogression lebte.
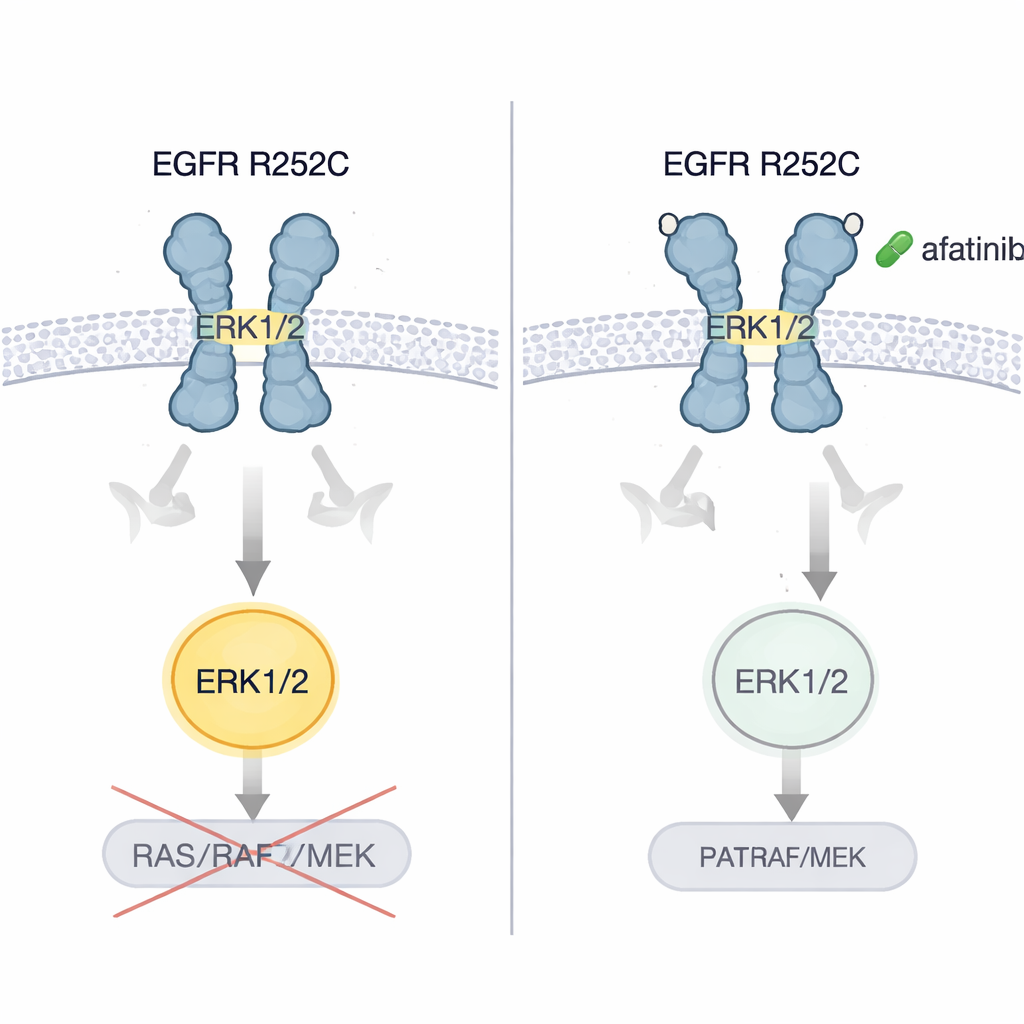
Was das für Patienten bedeutet
Diese Arbeit offenbart einen bisher unerkannte Mechanismus, wie eine krebsfördernde EGFR‑Mutation wirken kann: Indem sie zwei Rezeptoren außerhalb der Zelle zusammenklammert und sie in eine aktive Pose verdreht, schaltet sie ERK1/2 direkt ein, statt dem Lehrbuch‑Signalweg zu folgen. Für Nichtfachleute lautet die zentrale Erkenntnis: Nicht alle Mutationen im selben Gen verhalten sich gleich, und manche seltenen Veränderungen sprechen am besten auf spezifische vorhandene Medikamente an. EGFR R252C scheint Tumoren zu erzeugen, die besonders empfindlich auf Afatinib reagieren. Zwar stützt sich diese Schlussfolgerung derzeit auf einen detaillierten Patientenfall und umfangreiche Laborarbeiten, doch sie legt nahe, dass eine gezieltere Untersuchung äußerer EGFR‑Domänen‑Mutationen und eine sorgfältige Auswahl zielgerichteter Therapien neuen Patienten mit schwer behandelbaren Hirn‑ und Lungentumoren Hoffnung bieten könnten.
Zitation: Zhang, Y., Fei, Q., Li, Y. et al. An alternative EGFR activation by patient-derived R252C mutation promotes cancer progression. Nat Commun 17, 1902 (2026). https://doi.org/10.1038/s41467-026-68699-4
Schlüsselwörter: EGFR-Mutation, Gliom, Lungenkrebs, ERK-Signalgebung, Afatinib