Clear Sky Science · de
Verbesserung der Vorhersage polygenetischer Scores für unterrepräsentierte Gruppen durch Transferlernen
Warum Ihr DNA-Risikowert für Sie nicht funktionieren könnte
Genetische „Risikowerte“ werden zunehmend verwendet, um die Wahrscheinlichkeit einer Person abzuschätzen, an häufigen Erkrankungen wie Diabetes, Herzkrankheiten oder Bluthochdruck zu erkranken. Die meisten dieser Scores wurden jedoch mithilfe von DNA-Daten von Menschen mit europäischer Abstammung erstellt. Daher sagen sie für Menschen aus anderen Herkunftsgruppen oft unzuverlässig voraus, was Bedenken hinsichtlich Fairness und Nützlichkeit in der klinischen Praxis aufwirft. Diese Studie stellt eine einfache Frage: Können wir das, was wir aus großen europäischen Datensätzen gelernt haben, wiederverwenden, um bessere, gerechtere genetische Scores für unterrepräsentierte Gruppen zu erstellen — ohne die Rohdaten einzelner Personen weiterzugeben?
Von Millionen DNA-Markern zu einem einzigen Risikowert
Ein polygenetischer Score ist wie ein Zeugnis, das die kleinen Effekte vieler genetischer Marker über das Genom aufsummiert. Jeder Marker erhält ein Gewicht, das widerspiegelt, wie stark er mit einem Merkmal assoziiert ist, basierend auf großen genetischen Studien. Wenn diese Studien überwiegend Europäer einbeziehen, funktioniert der resultierende Score in der Regel am besten für Europäer. Unterschiede in der genetischen Ausstattung — wie häufig bestimmte DNA-Varianten sind und wie sie gemeinsam vererbt werden — bedeuten, dass dieselben Gewichte in afrikanisch-amerikanischen, hispanischen und anderen Populationen oft nicht zutreffen. Gleichgroße Datensätze für jede Gruppe zu sammeln ist teuer und zeitaufwendig, daher griffen die Autorinnen und Autoren zu einer Machine-Learning-Strategie namens Transferlernen: Statt in jeder Population neu anzufangen, verfeinern sie ein anderswo trainiertes Modell.
Wissen ausleihen, ohne Rohdaten zu teilen
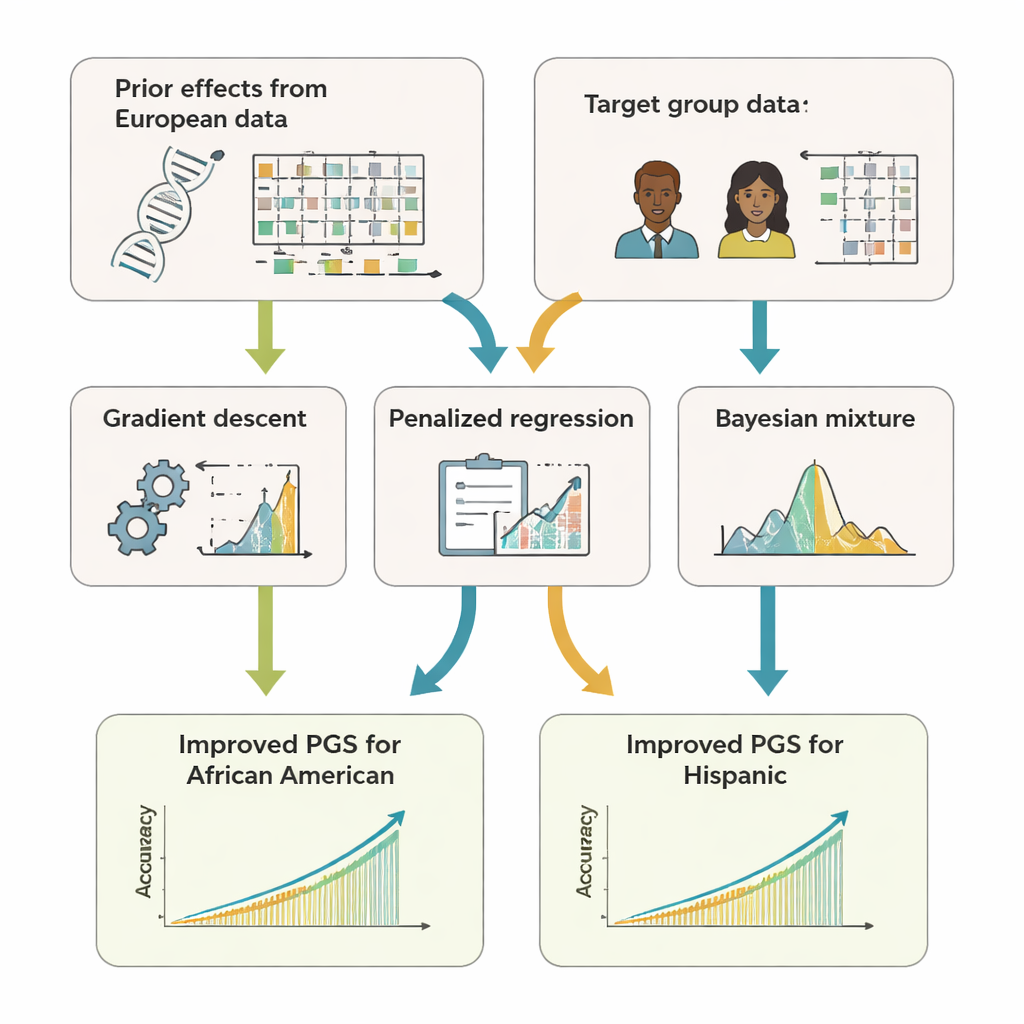
Das Team entwickelte GPTL, ein Open-Source-R-Paket, das drei Transferlernansätze für genetische Scores implementiert. Alle drei beginnen mit bestehenden Schätzungen der DNA-Effekte, die in einem großen Datensatz europäischer Abstammung gewonnen wurden, und passen diese Schätzungen dann behutsam mit Daten aus einer Zielgruppe wie Afroamerikanern oder Hispanics an. Eine Methode verändert die europäischen Gewichte schrittweise mittels Gradientenabstieg und stoppt früh, bevor sie diese vollständig überschreibt. Eine zweite Methode, die penalized Regression, zieht die neuen Schätzungen aktiv in Richtung der ursprünglichen Werte, es sei denn, die Zielgruppendaten liefern starke Hinweise auf Abweichungen. Das dritte Verfahren, ein Bayessches Mischmodell, erlaubt jedem DNA-Marker, unter mehreren Informationsquellen zu wählen — etwa verschiedenen Abstammungsgruppen oder einer "Kein-Effekt"-Option — und gewichtet sie entsprechend, wie gut sie die Zielgruppendaten erklären.
Die Methoden auf die Probe stellen
Um die Leistungsfähigkeit dieser Ansätze zu prüfen, nutzten die Autorinnen und Autoren sowohl Computersimulationen als auch reale Daten von Hunderttausenden Freiwilligen aus dem UK Biobank und dem US-Forschungsprogramm All of Us. Sie konzentrierten sich auf afroamerikanische und hispanische Teilnehmende als Zielgruppen und verwendeten Daten europäischer Abstammung als Hauptquelle für Vorinformationen. Bei 11 untersuchten Merkmalen — darunter Körpergröße, Body-Mass-Index, Blutlipide, Blutdruck und Nierenmarker — sagten die transfergelernten Scores durchweg besser voraus als Scores, die nur innerhalb der Zielgruppe erstellt wurden oder unverändert aus europäischen Daten übernommen wurden. Häufig erreichten sie eine Genauigkeit, die mit oder leicht über der von komplexeren "multi-ancestry"-Methoden lag, die das Zusammenführen von Rohdaten aus mehreren Populationen erfordern. Entscheidend ist, dass GPTL nur Zusammenfassungsstatistiken — aggregierte Zahlen zu genetischen Effekten — benötigt, sodass Institutionen zusammenarbeiten können, ohne individuelle genetische Datensätze offenzulegen.
Wenn mehr DNA nicht immer besser ist
Die Forschenden untersuchten außerdem, wie man am besten auswählt, welche genetischen Marker einbezogen werden sollten. Entgegen der verbreiteten Annahme, dass die Verwendung aller verfügbaren Marker immer nützt, stellten sie fest, dass das Einbeziehen von Millionen sehr schwacher Signale für afroamerikanische und insbesondere hispanische Gruppen die Leistung tatsächlich verschlechtern kann — vor allem bei stark vereinfachten Darstellungen genetischer Korrelationen. Die Konzentration auf besser unterstützte Marker und die Nutzung reichhaltigerer Informationen darüber, wie Varianten gemeinsam vererbt werden, führten häufiger zu genaueren Scores. Die Studie zeigte außerdem, dass das Hinzufügen von Vorinformationen aus mehreren Abstammungsgruppen und die sorgfältige Modellierung von Unterschieden zwischen Populationen die Vorhersagen weiter verbesserten.
Was das für gerechtere genetische Risikovorhersagen bedeutet
Für nicht-europäische Populationen können heutige Standardgenetikscores deutlich schlechter abschneiden und damit gesundheitliche Ungleichheiten verschärfen. Diese Arbeit zeigt, dass Transferlernen — das kluge Verfeinern auf europäischer Basis entwickelter Scores mit moderaten Datensätzen aus unterrepräsentierten Gruppen — einen großen Teil dieser Lücke schließen kann. Praktisch bedeutet das, dass Gesundheitssysteme und Forschende genauere und gerechtere genetische Werkzeuge entwickeln können, ohne Rohdaten institutions- oder abstammungsübergreifend zusammenzuführen, was Datenschutzbedenken verringert. Während keine einzelne Methode für jedes Merkmal und jede Population optimal sein wird, zeigt das GPTL-Toolkit, dass technisch gesehen gerechtere genetische Vorhersagen erreichbar sind, wenn wir vergangene Modelle nicht als feste Endprodukte, sondern als Ausgangspunkte betrachten, die für alle angepasst werden können.
Zitation: Wu, H., Pérez-Rodríguez, P., Boehnke, M. et al. Improving polygenic score prediction for underrepresented groups through transfer learning. Nat Commun 17, 1973 (2026). https://doi.org/10.1038/s41467-026-68696-7
Schlüsselwörter: polygenetische Risikowerte, Transferlernen, genetische Vorhersage, gesundheitliche Ungleichheiten, Populationsgenetik