Clear Sky Science · de
ProteoAutoNet: Hochdurchsatz-Analyse ko-elutierter Proteine mit Robotik und maschinellem Lernen
Warum das Verständnis von Proteinpartnerschaften wichtig ist
Innerhalb jeder Zelle arbeiten Proteine selten allein. Sie schließen sich in wechselnden Allianzen zusammen, um Strukturen aufzubauen, DNA zu kopieren, beschädigte Teile zu entfernen und Wachstum zu ermöglichen. Viele Krebsarten kapern diese Partnerschaften, doch ihre detaillierte Kartierung ist bisher langsam und mühsam gewesen. Diese Studie präsentiert ProteoAutoNet, ein von Robotik und maschinellem Lernen unterstütztes System, das die Entdeckung von Proteinpartnerschaften in Zellen deutlich beschleunigt und zeigt, wie dieser Ansatz verborgene Schwachstellen bei Schilddrüsenkrebsen aufdecken kann.
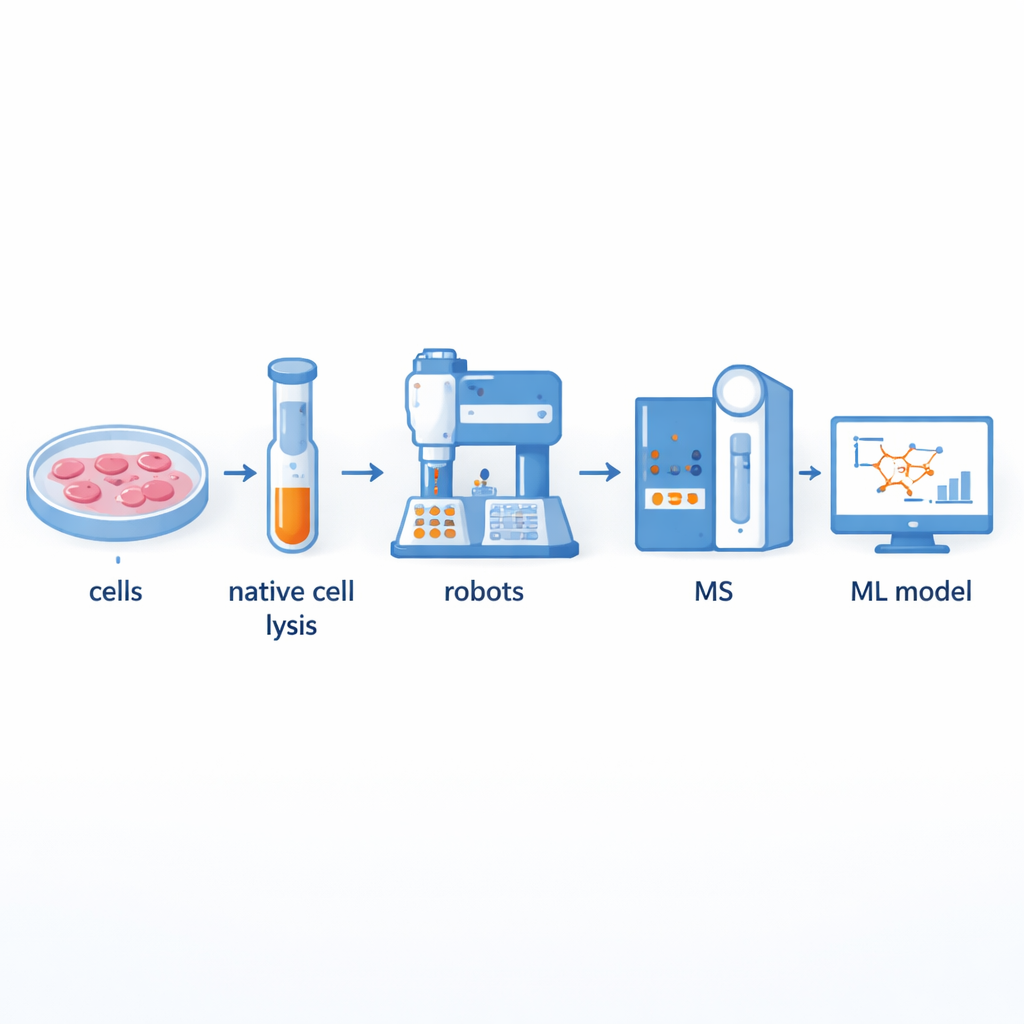
Aufbau einer schnelleren Proteinfabrik für Partnerschaften
Traditionell trennen Forschende große Proteinkomplexe mittels Co-Fraktionierungs-Massenspektrometrie und identifizieren anschließend deren Bestandteile. Obwohl diese Methode leistungsfähig ist, ist sie arbeitsintensiv und hat eine geringe Durchsatzrate: die manuelle Vorbereitung von Hunderten von Fraktionen kann viele Tage dauern. Die Autorinnen und Autoren bauten eine robotikgestützte Plattform, die den Großteil dieses Workflows automatisiert. Zellinhalte werden zunächst schonend aufgeschlossen, damit natürliche Proteinkomplexe intakt bleiben, und dann durch größenbasierte Säulen geführt, um sie in Dutzende Fraktionen zu trennen. Flüssigkeitshandhabungsroboter und Roboterarme übernehmen anschließend das Hinzufügen von Reagenzien, die Verdauung der Proteine in kleinere Peptide, die Aufreinigung der Proben und die Übergabe an ein Massenspektrometer zur Messung. Dieses System kann in nur zwei bis drei Tagen bis zu 540 Fraktionen aus mehreren Schilddrüsenzelllinien verarbeiten und verdoppelt damit etwa den Durchsatz im Vergleich zu früheren halbautomatischen Systemen.
Roboter, die nicht nur schneller, sondern auch zuverlässiger sind
Geschwindigkeit allein reicht nicht, wenn die Ergebnisse verrauscht oder inkonsistent sind. Das Team prüfte sorgfältig, ob die robotische Pipeline die Qualität der traditionellen manuellen Verarbeitung erreicht oder übertrifft. Mithilfe von Qualitätskontrollproben zeigten sie, dass das automatisierte System regelmäßig nahezu 3.000 Proteine pro Schilddrüsenzelllinie identifizierte, mit hoher Übereinstimmung zwischen Replikaten und starker Übereinstimmung in den gemessenen Proteinmengen. Beim direkten Vergleich von robotischer und manueller Verarbeitung derselben Proben detektierten beide Ansätze eine ähnliche Anzahl von Proteinen, doch die robotische Methode zeigte eine etwas geringere Variation in den Zählungen und stabilere Messungen der Proteinabundanzen. Das bedeutet, dass die neue Plattform nicht nur Zeit und Arbeitsaufwand spart, sondern auch reproduzierbarere Experimente ermöglicht — eine wichtige Voraussetzung für umfangreiche Studien und klinische Anwendungen.
Computern beibringen, sinnvolle Verbindungen zu erkennen
Selbst mit schnellen Geräten bleibt eine zentrale Herausforderung bestehen: zu entscheiden, welche Proteine tatsächlich interagieren und welche nur zufällig zusammen auftauchen. Zur Lösung kombinierten die Autorinnen und Autoren kuratierte Protein-Komplex-Datenbanken mit einem maschinellen Lernmodell auf Basis des XGBoost-Algorithmus. Zunächst bereinigten und fusionierten sie drei große Ressourcen zu Proteinkomplexen und erhielten so 96.635 bekannte Protein–Protein-Interaktionen. Sie verwendeten Profile dazu, wie Proteine über die Fraktionen hinweg erscheinen, als Eingangsmerkmale, und markierten Paare als wahrscheinliche Partner oder Nicht-Partner anhand der Datenbanken. Da echte, hochzuverlässige Partnerschaften relativ selten sind, setzten sie eine gezielte Datenaugmentationsstrategie ein: Sie erzeugten viele leicht veränderte Versionen bekannter positiver Beispiele, um dem Modell beizubringen, robuste Muster zu erkennen statt spezifische Signalverläufe auswendig zu lernen. Trainiert an Zehnern von Millionen solcher Beispiele aus drei Schilddrüsenzelllinien erzielte das Modell starke Leistungen und reihte wahre Interaktionen sowohl in internen Tests als auch in einer unabhängigen Validierungszelllinie deutlich über dem Zufallsniveau ein.
Neue Einblicke in die Maschinerie von Krebszellen
Mit diesem Workflow kartierten die Forschenden Interaktionsnetzwerke in einer normalen Schilddrüsenzelllinie und zwei krebsartigen Linien: einer papillären Schilddrüsenkarzinomlinie und einer follikulären Karzinomlinie, die in die Lunge metastasieren kann. Insgesamt identifizierten sie in diesen Zellen über 25.000 wahrscheinliche Proteininteraktionen und fanden starke Signale von bekannten zellulären Maschinerien wie Ribosomen (die Proteine bauen) und Proteasomen (die Proteine abbauen), was bestätigt, dass die Methode etablierte biologische Strukturen erfasst. Durch den Vergleich der Krebszellen mit der normalen Linie entdeckten sie Netzwerke, die in der Krankheit hochreguliert waren. In den metastatischen follikulären Karzinomzellen waren sowohl Proteasomkomponenten als auch ein Chaperon-Komplex namens Prefoldin deutlich stärker vernetzt und vermehrt vorhanden. Mehrere Prefoldin-Untereinheiten waren zuvor mit anderen Krebsarten in Verbindung gebracht worden, doch globale Proteinsurveys hatten ihr koordiniertes Verhalten bei Schilddrüsenkrebs möglicherweise übersehen, weil diese Proteine streng durch Abbau reguliert werden. Der Co-Fraktionierungsansatz offenbarte ihre koordinierten Veränderungen auf Komplexebene.
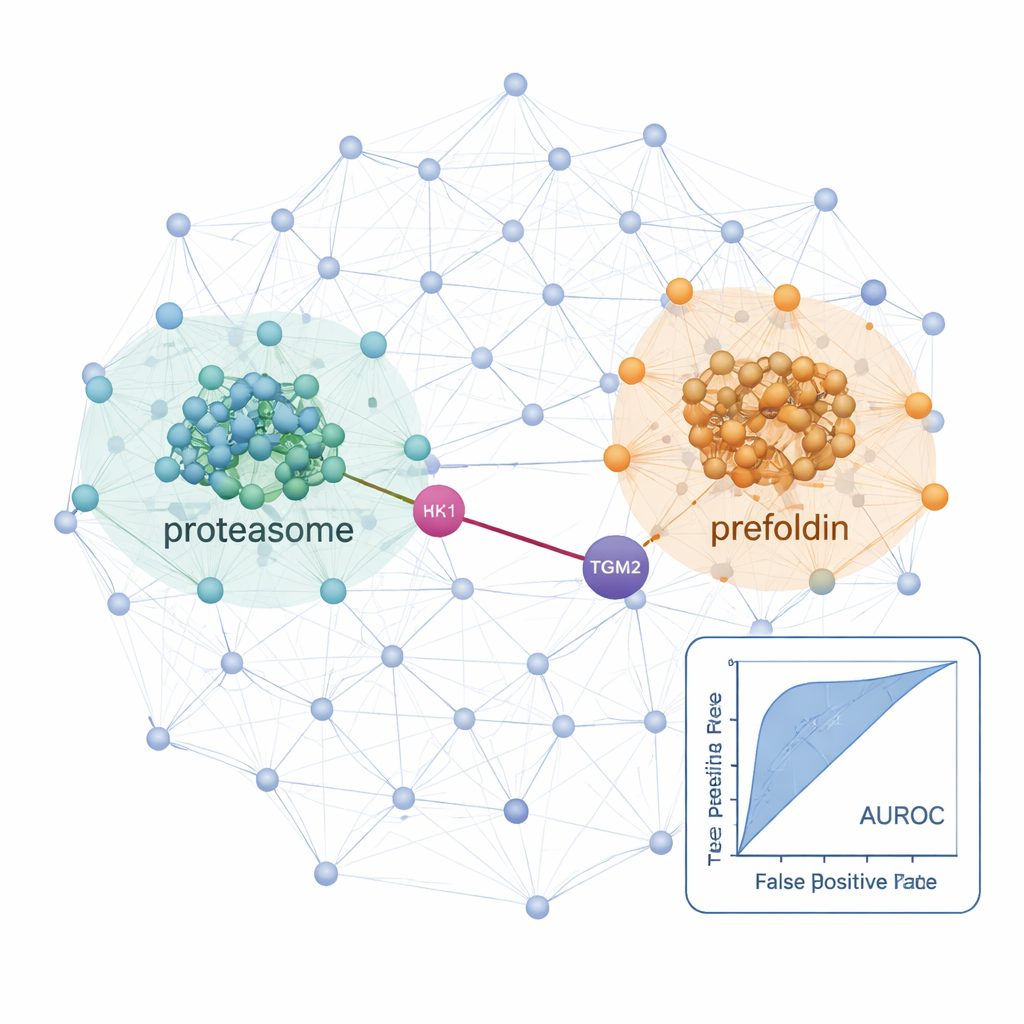
Verborgene Verbindungen, die künftige Behandlungen leiten könnten
Die Studie hob auch spezifische Interaktionen hervor, die für das Wachstum und die Ausbreitung von Schilddrüsenkrebsen relevant sein könnten. Ein Beispiel ist eine vorhergesagte Partnerschaft zwischen HK1, einem Enzym, das den wichtigsten Zuckerabbauweg der Zelle startet, und TGM2, einem Protein, das für Invasion und Metastasierung in Schilddrüsentumoren bekannt ist. Diese HK1–TGM2-Verbindung, die in bestehenden Interaktionsdatenbanken fehlt, wurde durch strukturelles Modellieren gestützt und zeigte sich besonders aktiv in der papillären Karzinomlinie, was nahelegt, dass metabolische Umprogrammierung und invasives Verhalten physisch verknüpft sein könnten. Zusammenfassend zeigt ProteoAutoNet, wie die Kombination von Robotik und maschinellem Lernen langsame, expertengesteuerte Kartierungen von Proteinnetzwerken in einen skalierbareren Prozess verwandeln kann. Für nicht Spezialisten lautet die Kernbotschaft, dass diese Technologie sowohl breite Verschiebungen in der zellulären Maschinerie als auch unerwartete Proteinpartnerschaften aufdecken kann, die eines Tages Ärzten helfen könnten, besser vorherzusagen, welche Schilddrüsenkrebse aggressiv sind, und neue Therapieziele zu identifizieren.
Zitation: Lyu, M., Hu, P., Zhang, G. et al. ProteoAutoNet: high-throughput co-eluted protein analysis with robotics and machine learning. Nat Commun 17, 1949 (2026). https://doi.org/10.1038/s41467-026-68686-9
Schlüsselwörter: Proteininteraktionen, Massenspektrometrie, maschinelles Lernen in der Biologie, Schilddrüsenkrebs, Proteasom und Prefoldin