Clear Sky Science · de
NeoPrecis: Verbesserung der Vorhersage von Immuntherapieansprechen durch Integration qualifizierter immunogener und klonalitätsbewusster Neoantigen-Landschaften
Warum manche Krebsarten auf Immuntherapie ansprechen und andere nicht
Die Immuntherapie hat die Krebsbehandlung revolutioniert, doch viele Patientinnen und Patienten profitieren weiterhin nicht, und einige erleiden schwere Nebenwirkungen. Eine zentrale Frage ist, warum manche Tumoren vom Immunsystem erkannt und zerstört werden, während andere unentdeckt bleiben. Diese Studie stellt NeoPrecis vor, eine rechnerische Methode, die die „Signale“, die Tumoren dem Immunsystem senden — sogenannte Neoantigene — genauer untersucht und diese Informationen nutzt, um besser vorherzusagen, welche Patientinnen und Patienten wahrscheinlich auf moderne Immuntherapien ansprechen.
Neue Signale auf Krebszellen
Krebszellen sammeln DNA‑Mutationen an, die die von ihnen produzierten Proteine verändern können. Kleine Fragmente dieser veränderten Proteine, bekannt als Neoantigene, können an der Zelloberfläche präsentiert und von T‑Zellen, den Effektorzellen des Immunsystems, als fremd erkannt werden. Jahrelang haben Ärztinnen, Ärzte und Forschende auf grobe Maße wie die Tumormutationslast — also die Gesamtzahl der Mutationen — gesetzt, um abzuschätzen, wie wahrscheinlich ein Tumor auf Checkpoint‑Inhibitoren anspricht. Das ist jedoch ein ungenaues Instrument: Nicht jede Mutation erzeugt ein sichtbares oder attraktives Ziel für T‑Zellen, und Tumoren können aus einem Flickenteppich vieler unterschiedlicher Zelluntertypen bestehen. NeoPrecis wurde entwickelt, um über das bloße Zählen von Mutationen hinauszugehen und stattdessen zu bewerten, wie viele davon tatsächlich vielversprechende Ziele im gesamten Tumor darstellen.
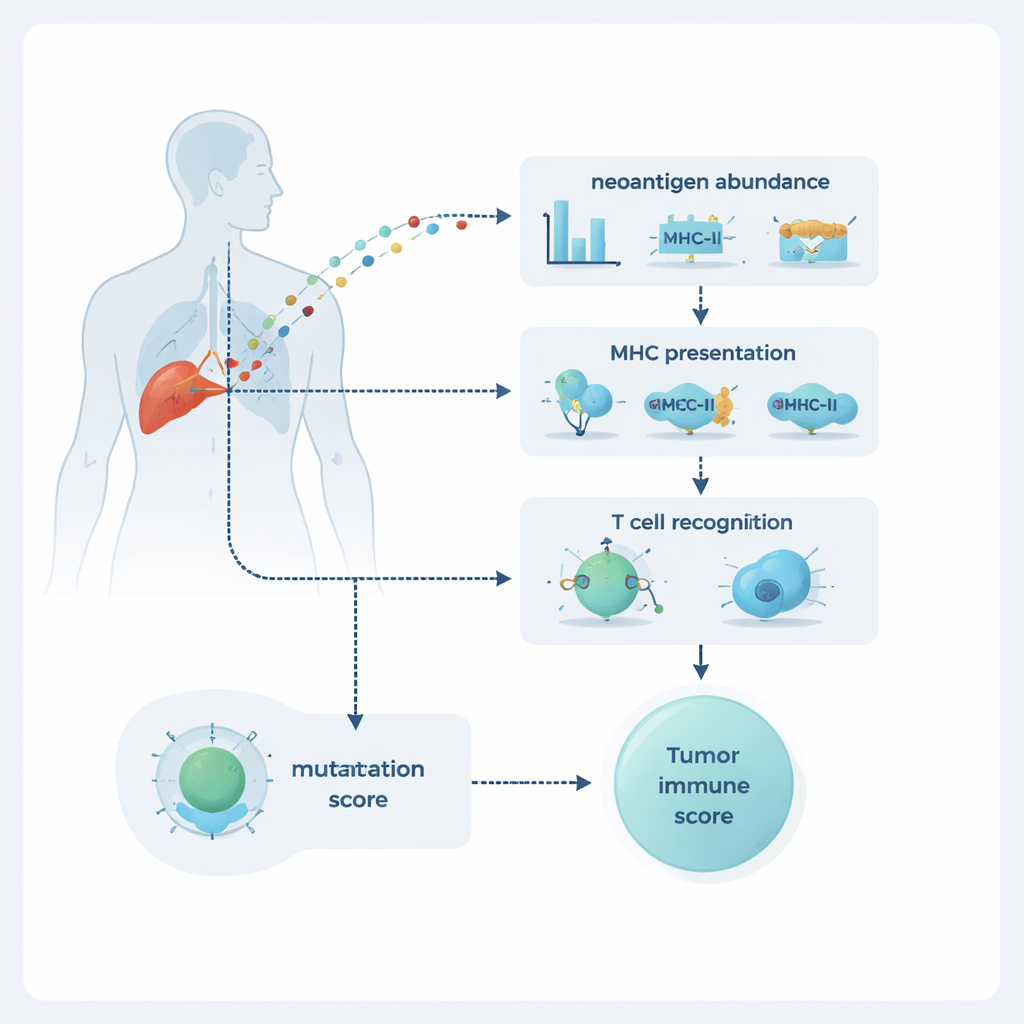
Drei Schlüsselkomponenten gleichzeitig betrachten
NeoPrecis bewertet jede Mutation entlang dreier Dimensionen: wie häufig sie im Tumor vorkommt, ob sie wahrscheinlich an der Zelloberfläche präsentiert wird und wie wahrscheinlich sie von T‑Zellen erkannt wird. Die Häufigkeit wird aus DNA‑ und RNA‑Sequenzdaten geschätzt, die zeigen, wie verbreitet die Mutation ist und wie stark sie exprimiert wird. Die Präsentation wird über die Bindung an Moleküle der MHC‑Klasse I und II modelliert, die wie Schautafeln Peptidfragmente T‑Zellen zeigen. Der innovativste Teil ist die Komponente zur T‑Zell‑Erkennung, NeoPrecis‑Immuno. Dieses Modell lernt aus großen Datenbanken bekannter T‑Zell–Peptid‑Interaktionen, wie stark sich ein mutiertes Fragment von seinem normalen Gegenstück in einer für die T‑Zell‑Erkennung relevanten Weise unterscheidet, und berücksichtigt dabei auch die spezifischen MHC‑Varianten einer Person.
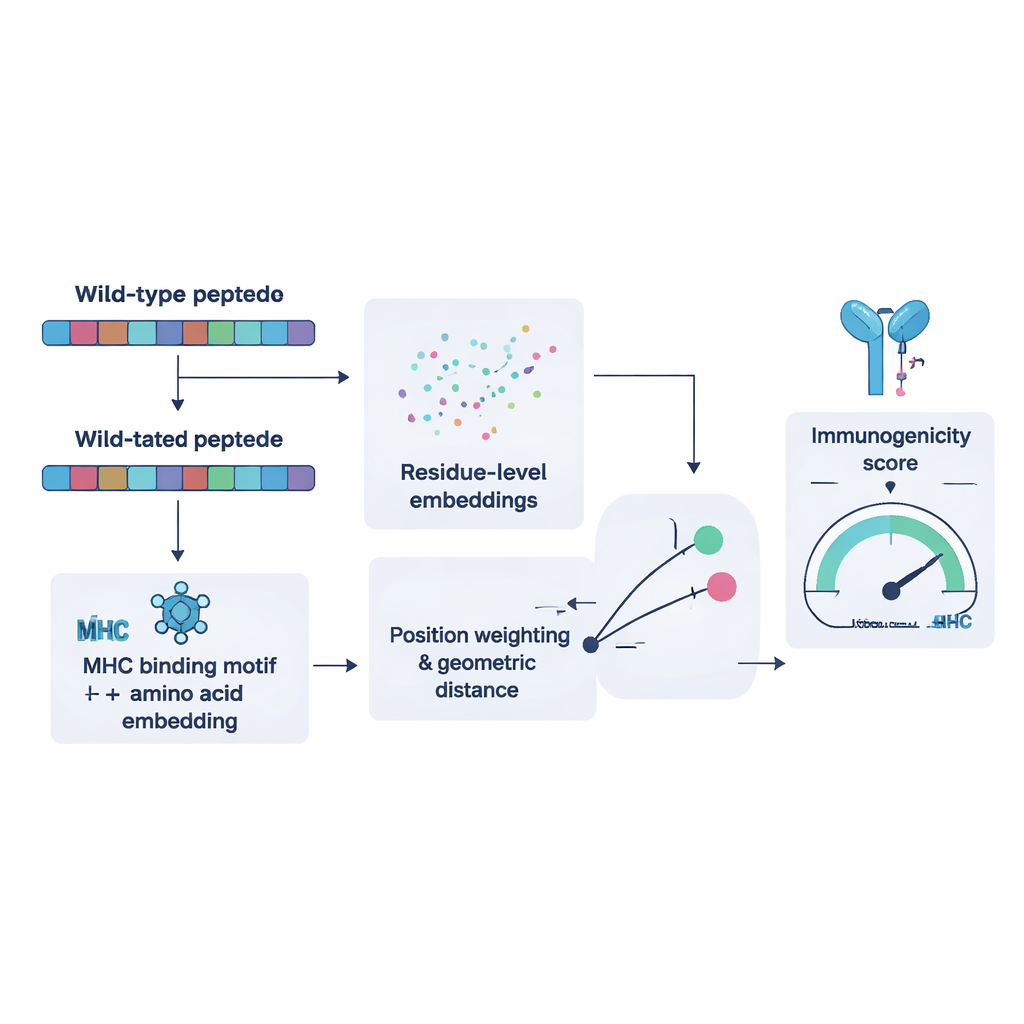
Einem Computer beibringen, was T‑Zellen „sehen“
Zum Trainieren von NeoPrecis‑Immuno stellten die Forschenden zunächst Tausende von Beispielen zusammen, in denen dieselbe T‑Zelle mehrere ähnliche Peptide erkennt und andere nicht. Diese Beispiele nutzten sie, um dem Modell beizubringen, dass mutierte Fragmente, die dem normalen sehr ähnlich sind, weniger wahrscheinlich eine Immunreaktion auslösen, weil solche selbstähnlichen Ziele während der T‑Zell‑Entwicklung meist aussortiert werden. Das Modell stellt jedes Peptid als Punkt in einem mathematischen Raum dar, der sowohl von seiner Aminosäuresequenz als auch von den Bindungspräferenzen der MHC‑Moleküle der jeweiligen Person geprägt ist. Anschließend misst es, wie weit das mutierte Peptid vom Original entfernt liegt. Größere, motiv‑informierte Distanzen entsprechen einer höheren Wahrscheinlichkeit, immunogen zu sein. Im Vergleich mit bestehenden Werkzeugen auf unabhängigen Krebsdatensätzen erreichte NeoPrecis‑Immuno ähnliche oder bessere Leistungen, besonders bei der Arbeit mit MHC Klasse II, die Helfer‑T‑Zellen präsentiert, die die antitumorale Antwort unterstützen und aufrechterhalten.
Von einzelnen Mutationen zum gesamten Tumor
Einzelne Mutationen erzählen nur einen Teil der Geschichte; auch ihre Verteilung im Tumor ist wichtig. Manche Mutationen sind „klonal“ und in nahezu jeder Krebszelle zu finden, andere sind „subklonal“ und nur in bestimmten Bereichen vorhanden. NeoPrecis erstellt eine „Neoantigen‑Landschaft“, indem es seine Immunogenitätswerte über die Mutationen aufsummiert und Informationen darüber schichtet, zu welchen Subklonen sie gehören und wie häufig diese Subklone sind. So entstehen Tumor‑Level‑Scores, die Tumoren hervorheben, die reich an starken, weit verbreiteten Neoantigenen sind — insbesondere solchen, die sowohl auf MHC Klasse I als auch Klasse II präsentiert werden können und potenziell koordinierte Helfer‑ und Killer‑T‑Zell‑Reaktionen auslösen. In Patientengruppen mit Melanom und nicht‑kleinzelligem Lungenkarzinom, die mit Checkpoint‑Inhibitoren behandelt wurden, trennten diese NeoPrecis‑basierten Scores Responder besser von Nicht‑Respondern als standardmäßige Mutationszählungen und waren besonders hilfreich bei komplexen, heterogenen LungenTumoren.
Was das für Patientinnen und Patienten bedeutet
Für Patientinnen und Patienten liegt das Versprechen von NeoPrecis in einer präziseren Zuordnung von Immuntherapien an diejenigen, die am ehesten profitieren, sowie in einem klareren Verständnis, warum manche Tumoren Therapie widerstehen. Indem der Fokus auf Qualität und Verteilung der Neoantigene — nicht nur auf deren Menge — gelegt wird, hilft das Framework zu erklären, warum Tumoren mit ähnlichen Mutationslasten sehr unterschiedlich reagieren können. In Zukunft könnten solche detaillierten Karten der immun‑sichtbaren Landschaft eines Tumors nicht nur die Anwendung von Checkpoint‑Inhibitoren leiten, sondern auch die Entwicklung personalisierter Krebsimpfstoffe unterstützen, die die wirksamsten und am weitesten verbreiteten Ziele in jedem individuellen Tumor priorisieren.
Zitation: Lee, KH., Sears, T.J., Zanetti, M. et al. NeoPrecis: enhancing immunotherapy response prediction through integration of qualified immunogenicity and clonality-aware neoantigen landscapes. Nat Commun 17, 1966 (2026). https://doi.org/10.1038/s41467-026-68651-6
Schlüsselwörter: Krebsimmuntherapie, Neoantigene, Tumorheterogenität, Checkpoint-Inhibitoren, computationale Onkologie