Clear Sky Science · de
Abschätzung der Proteinfaltungsstabilität mit expliziter Berücksichtigung ungefalteter Zustände
Warum Proteinstabilität wichtig ist
Jedes Protein in Ihrem Körper ist eine winzige molekulare Maschine, die sich in eine präzise dreidimensionale Gestalt falten muss, um korrekt zu funktionieren. Ist diese Faltung zu labil, kann das Protein fehlgehen, verklumpen oder gar nicht erst produziert werden – Probleme, die mit Krankheiten sowie mit dem Scheitern bei der Herstellung proteinbasierter Medikamente und Enzyme verbunden sind. Die Messung, wie stabil ein Protein im Labor ist, ist zeitaufwändig und schwierig, weshalb Forscher nach computergestützten Methoden suchen, die zuverlässig allein aus der Sequenz vorhersagen können, wie leicht ein Protein entfaltet wird.
Ein neuer Blick auf gefaltete und ungefaltete Proteine
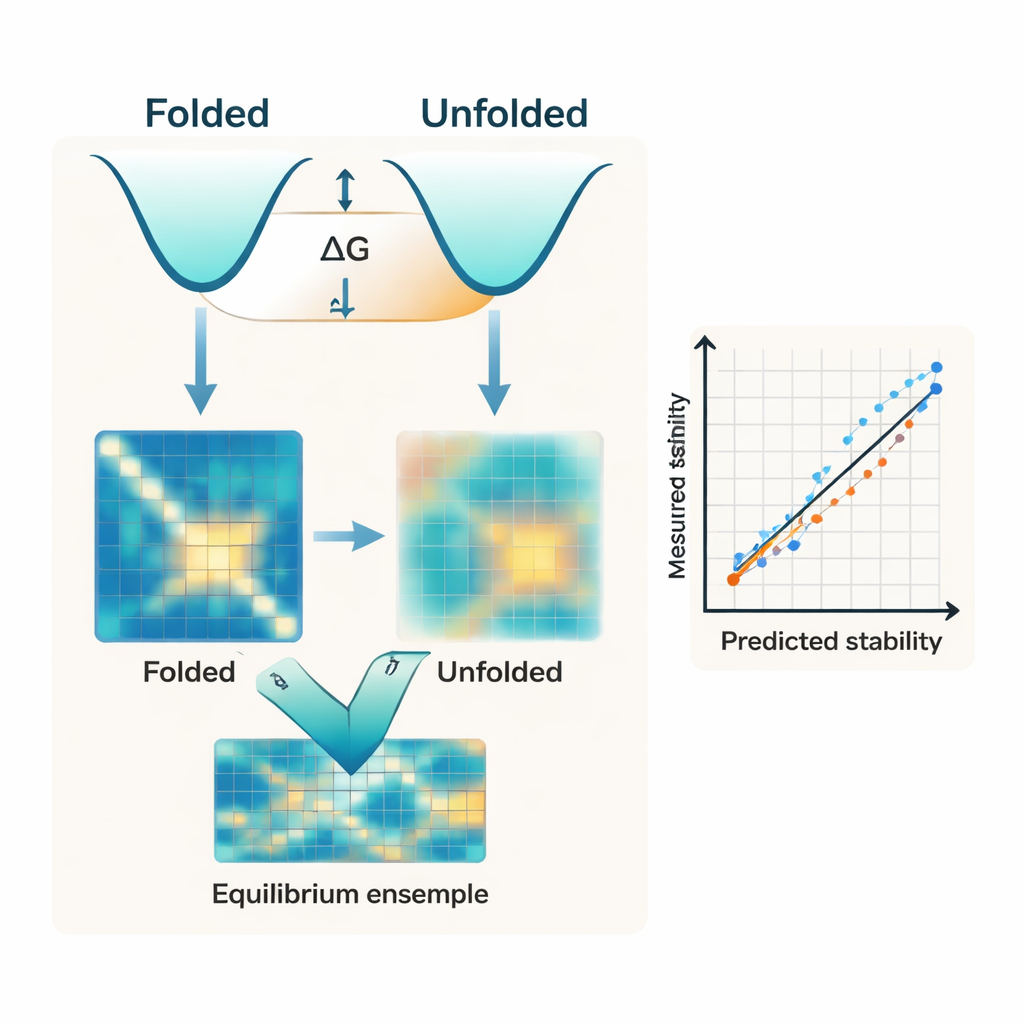
Die meisten modernen Algorithmen konzentrieren sich fast ausschließlich auf die gefaltete Form eines Proteins. Sie beginnen oft mit einer KI‑prognostizierten Struktur, etwa von AlphaFold, und behandeln diese einzelne Struktur als den Hauptfaktor für Stabilität. Stabilität ist jedoch tatsächlich die Energiedifferenz zwischen zwei breiten Ensembles: dem kompakten gefalteten Zustand und den vielen flexiblen Konformationen, die den ungefalteten Zustand ausmachen. Die Autor:innen argumentieren, dass die Vernachlässigung der ungefalteten Seite dieses Gleichgewichts ein zentraler Grund dafür ist, dass bestehende Werkzeuge Schwierigkeiten haben, experimentelle Messungen der Faltungsfreienergie, bekannt als ΔG, genau vorherzusagen.
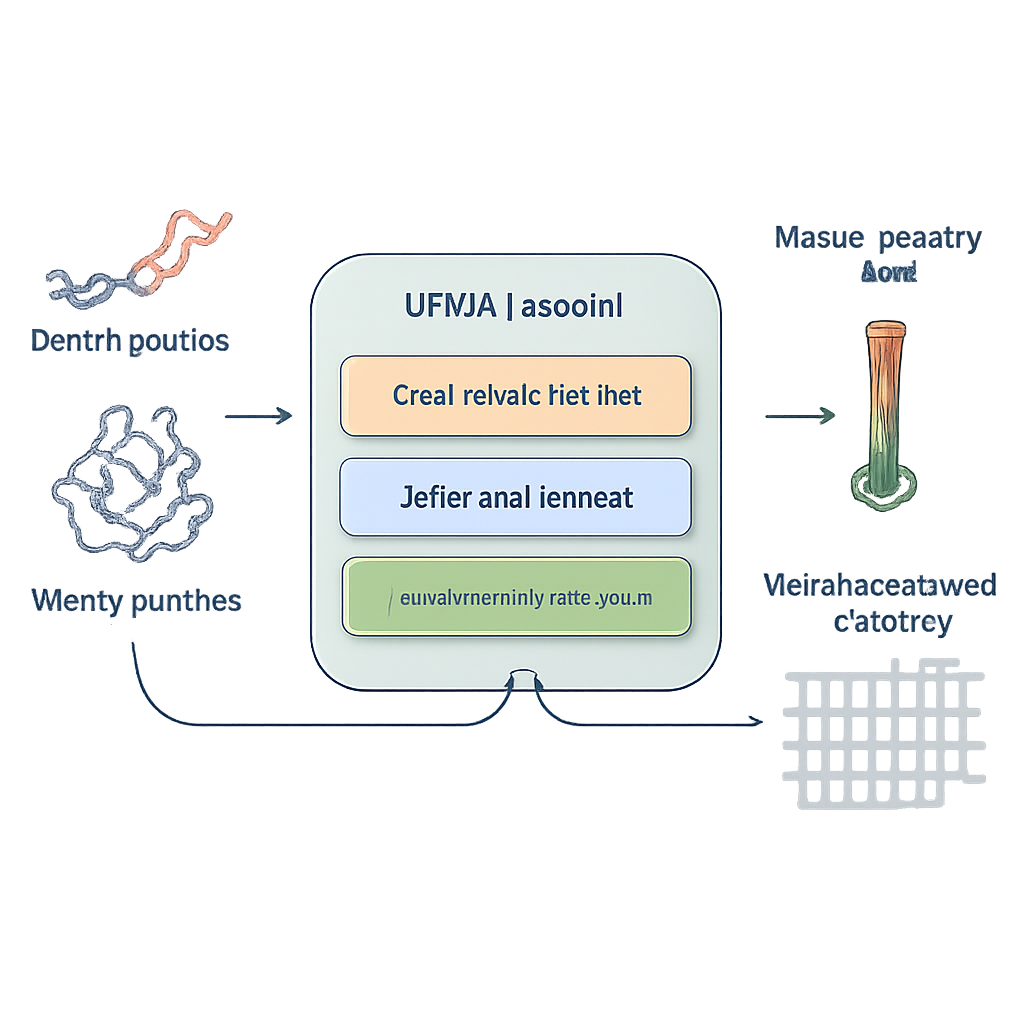
Ein neues Modell, das beide Zustände lernt
Die Forschenden stellen IFUM vor, ein System des tiefen Lernens, das darauf ausgelegt ist, ΔG abzuschätzen und gleichzeitig zu lernen, wie das Verhältnis von gefalteten und ungefalteten Zuständen für jedes Protein aussieht. Anstatt den ungefalteten Zustand als vagen Hintergrund zu behandeln, nutzt IFUM Konzepte aus der Polymerphysik, um ihn als „random coil“ darzustellen, und kodiert sowohl gefaltete als auch ungefaltete Zustände als Abstandskarten zwischen Aminosäurepaaren. Das Modell bezieht Informationen aus leistungsfähigen, vortrainierten Sequenz‑ und Struktur‑Netzwerken und sagt gemeinsam die Gesamtstabilität und eine Wahrscheinlichkeitskarte voraus, die beschreibt, wie groß der Anteil der gefalteten gegenüber der ungefalteten Population an jedem Restpaar ist. Das Training an einem sehr großen Datensatz aus kleinen, experimentell charakterisierten Proteinen und bekannten ungeordneten Proteinen hilft IFUM, sowohl gut strukturierte als auch flexible Sequenzen zu erkennen.
Bessere Zahlen und breitere Abdeckung von Mutationen
Getestet an einem sorgfältig kontrollierten Datensatz kleiner Proteine sagt IFUM experimentelle ΔG‑Werte mit geringerer Fehlerabweichung und höherer Korrelation vorher als frühere KI‑basierte Methoden, die nur auf der gefalteten Struktur oder auf mit Sequenzen trainierten Sprachmodellen beruhen. Entscheidend ist, dass das Modell auch eine große Vielfalt an Sequenzveränderungen handhabt. Es erfasst zuverlässig die Effekte von Einzel‑ und Doppelpunktmutationen sowie Einfügungen und Deletionen, die die Proteingröße verändern – Situationen, in denen viele bestehende Werkzeuge entweder vollständig versagen oder nicht dafür ausgelegt sind. Ein interner Vergleich zeigt, dass das Entfernen des Ziels, den ungefalteten Zustand zu modellieren, die Leistung deutlich verschlechtert, was unterstreicht, dass die explizite Modellierung des ungefalteten Ensembles nicht nur ein konzeptionelles Extra ist, sondern zentral für die Genauigkeit der Vorhersagen.
Vom Design‑Labor zur Praxisprüfung
Um zu prüfen, ob IFUM das reale Protein‑Engineering leiten kann, wenden die Autor:innen es auf drei anspruchsvolle Designaufgaben an: Stabilisierung von Interferon‑Lambda‑Proteinen, Umgestaltung des Immunsignalproteins IL‑10 und Verbesserung eines zuckermodifizierenden Enzyms namens UGT76G1. In allen drei Fällen stimmen IFUMs vorhergesagte Stabilitäten gut mit gemessenen Schmelzpunkten überein, die angeben, wie viel Wärme ein Protein aushält, bevor es entfaltet. Das Modell hilft auch dabei, Hunderte neu entworfener, rechnerisch erzeugter Proteine zu sichten, um diejenigen auszuwählen, die am ehesten falten und in Zellen löslich bleiben, und übertrifft dabei weit verbreitete Vertrauensmaße aus Struktur‑Vorhersagenetzwerken. Diese Ergebnisse deuten darauf hin, dass IFUM als praktischer „Stabilitätsfilter“ neben struktur‑basierten Prüfungen in modernen Protein‑Design‑Workflows eingesetzt werden kann.
Grenzen und zukünftige Richtungen
Wie jedes Modell hat auch IFUM seine Grenzen. Es ist hauptsächlich an kurzen, einkettigen, löslichen Proteinen trainiert, und seine absoluten Stabilitätswerte werden für deutlich größere Proteine oder solche mit ausgedehnten flexiblen Schleifen oder membranüberspannenden Regionen weniger vertrauenswürdig. Seine Beschreibung des ungefalteten Zustands bleibt ein vereinfachtes statistisches Modell und kein vollständig realistisches Abbild aller möglichen Konformationen. Dennoch zeigt der Ansatz, dass das Lehren einer KI, sowohl gefaltete als auch ungefaltete Ensembles zu berücksichtigen, zu verlässlicheren Stabilitätsabschätzungen führt. Für Nicht‑Expert:innen ist die zentrale Erkenntnis, dass IFUM uns näher bringt an die Möglichkeit, einem Computer mit quantitativer Zuverlässigkeit die Frage zu stellen: „Wird dieses Protein‑Design wirklich zusammenhalten?“, und damit die Entwicklung sichererer biologischer Medikamente und robusterer industrieller Enzyme zu beschleunigen.
Zitation: Lee, H., Cho, Y., Yun, J. et al. Protein folding stability estimation with explicit consideration of unfolded states. Nat Commun 17, 1883 (2026). https://doi.org/10.1038/s41467-026-68637-4
Schlüsselwörter: Proteinstabilität, Proteinfaltung, Tiefes Lernen, Protein‑Design, Mutationen