Clear Sky Science · de
Umfassender Benchmarking-Vergleich von ein- und multi-ethnischen polygenen Score-Methoden mit der PGS-hub-Plattform
Warum Ihr DNA‑Risikowert wichtig ist
Ärztinnen und Ärzte werden immer besser darin, unsere DNA zu lesen, um abzuschätzen, wer mit größerer Wahrscheinlichkeit häufige Erkrankungen wie Herzkrankheiten, Diabetes oder Schizophrenie entwickelt. Diese Schätzungen, polygene Scores genannt, fassen die winzigen Effekte vieler genetischer Varianten zu einer einzigen Zahl zusammen. Es gibt inzwischen jedoch viele konkurrierende Möglichkeiten, solche Scores zu berechnen, und sie funktionieren nicht für Menschen aus allen Abstammungshintergründen gleichermaßen gut. Diese Studie hatte zum Ziel, führende Methoden direkt zu vergleichen und einen Online‑Dienst, PGS‑hub, zu entwickeln, der Forschern erlaubt, diese Scores konsistent und einfach zu berechnen.
Eine Anlaufstelle für DNA‑Risikorechner
Die Autorinnen und Autoren schufen PGS‑hub, eine Webplattform, die einen Großteil der technischen Komplexität hinter polygenen Scores verbirgt. Nutzer laden Ergebnisdaten aus genetischen Studien hoch, die zusammenfassen, wie Millionen von DNA‑Markern mit einer Krankheit oder einem Merkmal zusammenhängen. Anschließend wählen sie den Abstammungshintergrund der Population, die sie interessiert – beispielsweise europäisch oder afrikanisch – und wählen aus einem Menü populärer Bewertungsmethoden. Im Hintergrund wandelt PGS‑hub die Eingaben in die richtigen Formate um, verwendet vorgefertigte Referenz‑Panels, die beschreiben, wie nahegelegene DNA‑Marker korrelieren, und startet zahlreiche Jobs auf einem Hochleistungsrechnersystem. Das Ergebnis ist eine kompakte Gewichtungsdatei, die auf individuelle Genome angewendet werden kann, um für jede Person einen Score zu erzeugen. 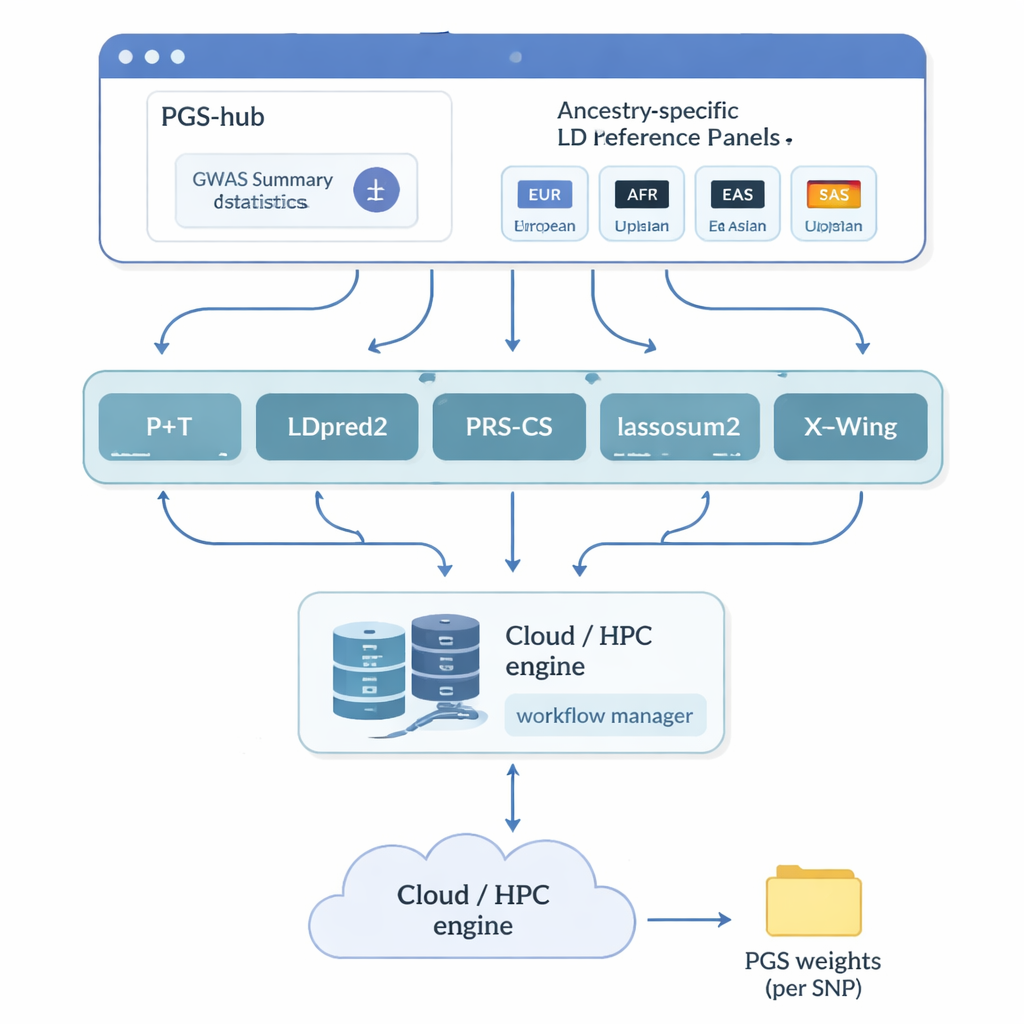
13 Methoden im Vergleich
Um herauszufinden, welche Ansätze am besten funktionieren, verglich das Team 13 moderne Methoden über 36 Erkrankungen und Merkmale in fast 380.000 Personen europäischer Abstammung und etwas mehr als 8.000 Personen afrikanischer Abstammung aus der UK Biobank. Sie bewerteten nicht nur, wie gut jeder Score vorhersagte, wer eine Krankheit hatte oder höhere Merkmalswerte zeigte, sondern auch, wie viel Rechenzeit und Arbeitsspeicher jede Methode benötigte. Bei Europäern lieferte eine Methode namens LDpred2 im Allgemeinen die genauesten Scores und übertraf oft die anderen deutlich. Einige Alternativen – lassosum2, PRS‑CS und SDPR – schnitten für viele Merkmale nahezu ebenso gut ab, während einige ältere Methoden zurückblieben. Für Merkmale wie Körpergröße oder Morbus Crohn erklärten die besten Scores einen beträchtlichen Anteil des genetischen Risikos; für andere, etwa Nierenfunktion, taten sich alle Methoden schwer, was auf schwächere zugrunde liegende genetische Signale hinweist.
Erkenntnisse für vielfältige Populationen und kombinierte Methoden
Ein zentrales Problem in der genetischen Vorhersage ist, dass in erster Linie an Europäern trainierte Methoden möglicherweise nicht gut auf Menschen mit anderen Abstammungen übertragbar sind. Als die Autorinnen und Autoren ihre Benchmarks mit genetischen Studien afrikanischer Abstammung wiederholten, schnitt jede Methode schlechter ab, was den Mangel an großen Studien in diesen Populationen verdeutlicht. Dennoch gehörten LDpred2 und SDPR tendenziell zu den besseren Optionen. Das Team untersuchte außerdem „Multi‑Ancestry“-Ansätze, die explizit Informationen über Populationen hinweg kombinieren. Hier übertraf eine relativ einfache Strategie – die lineare Kombination der besten abstammungsspezifischen LDpred2‑Scores zu einem einzigen LDpred2‑multi‑Score – komplexere multi‑ethnische Modelle wie PRS‑CSx und X‑Wing sowohl in europäischen als auch in afrikanischen Gruppen. Darüber hinaus zeigten die Autorinnen und Autoren, dass ein Ensemble, das die stärksten Scores mehrerer Methoden mischt, die Vorhersage über alle Merkmale hinweg weiter verbesserte, insbesondere bei hoch erblichen Erkrankungen wie Schizophrenie und koronarer Herzkrankheit. 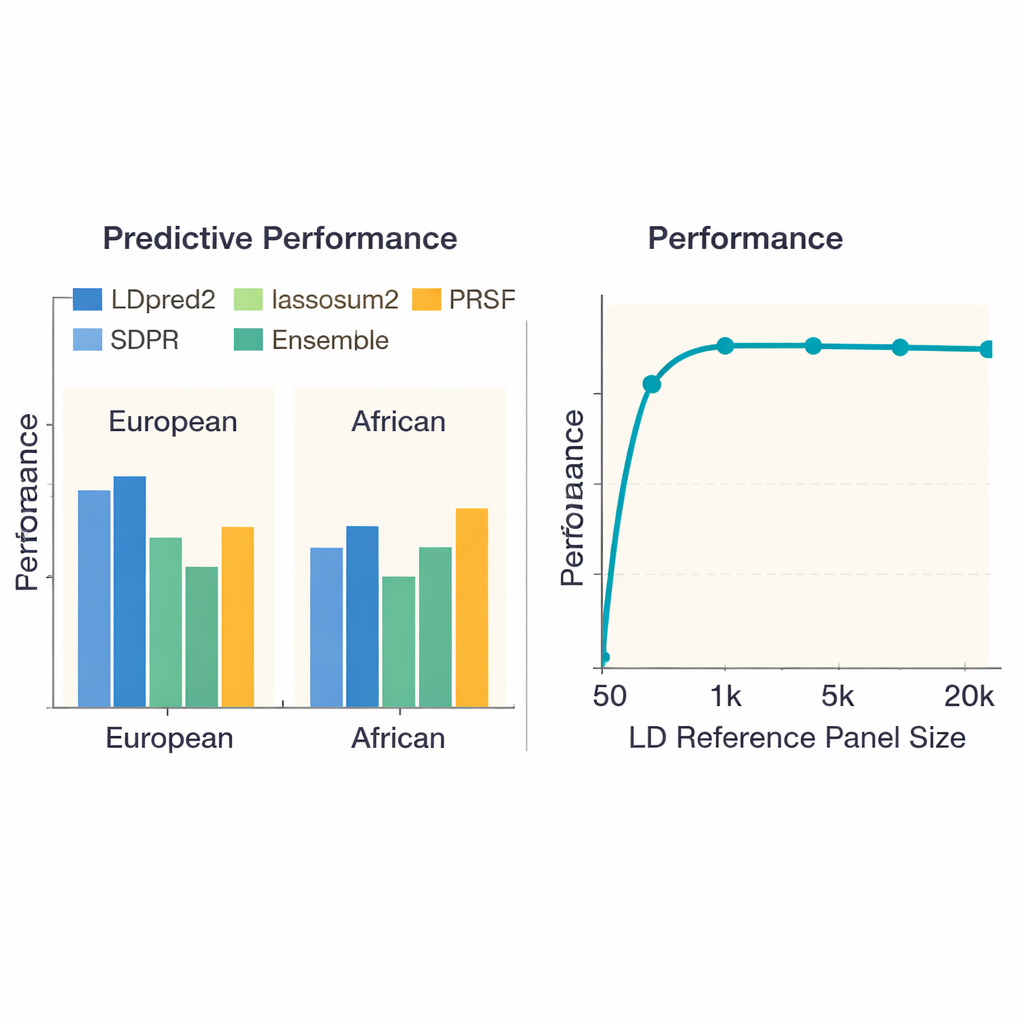
Wie Datenwahl und Rechenlimits Scores prägen
Die Studie untersuchte, wie die Größe des Referenz‑Panels – der Personengruppe, die verwendet wird, um zu lernen, wie nahegelegene DNA‑Marker kovariieren – die Leistung beeinflusst. Wenn dieses Panel sehr klein war (weniger als 1.000 Individuen), waren die Scores deutlich weniger genau. Mit einer Panelgröße von etwa 5.000 Personen verbesserte sich die Leistung stark und flachte dann ab, was darauf hindeutet, dass immer größere Panels abnehmende Erträge bringen. Überraschenderweise half das Hinzufügen von mehr DNA‑Markern nicht immer: Die Verwendung von etwa 6,6 Millionen Varianten verschlechterte in manchen Fällen die Vorhersagen gegenüber einer sorgfältig ausgewählten Menge von ungefähr 1,1 Millionen, wahrscheinlich weil zusätzliche Marker mehr Rauschen als nützliche Signale einbrachten. Die Autorinnen und Autoren dokumentierten auch große Unterschiede bei den Rechenkosten. Einfache Methoden wie grundlegendes Pruning‑und‑Thresholding waren pro Merkmal in weniger als einer Stunde fertig, während einige bayesianische Ansätze Hunderte von CPU‑Stunden erforderten – Informationen, die für große Projekte oder ressourcenbeschränkte Gruppen wichtig sind.
Was das für künftige DNA‑basierte Vorhersagen bedeutet
Für Nicht‑Spezialisten lautet die Kernbotschaft: Nicht alle DNA‑Risikowerte sind gleichwertig, und die Details ihrer Konstruktion beeinflussen stark, wer von ihnen profitiert. Diese Arbeit liefert praktische Orientierung: Methoden wie LDpred2 und gut konzipierte Ensembles liefern in großen europäischen Datensätzen tendenziell die zuverlässigsten Vorhersagen, und Multi‑Ancestry‑Kombinationen können komplexere populationsübergreifende Modelle übertreffen. Gleichzeitig unterstreicht der Genauigkeitsverlust bei Personen afrikanischer Abstammung den dringenden Bedarf an größeren und diverseren genetischen Studien. Indem viele Methoden in einer einzigen, standardisierten Online‑Plattform gebündelt werden, senkt PGS‑hub die Hürde für Forschende weltweit, polygene Scores zu erzeugen und zu vergleichen – ein wichtiger Schritt, um solche Scores fair und effektiv in der Medizin einzusetzen.
Zitation: Chen, X., Wang, F., Zhao, H. et al. Comprehensive benchmarking single and multi ancestry polygenic score methods with the PGS-hub platform. Nat Commun 17, 2014 (2026). https://doi.org/10.1038/s41467-026-68599-7
Schlüsselwörter: polygene Scores, genetische Risikoabschätzung, PGS-hub-Plattform, Multi‑Ancestry-Genomik, UK Biobank