Clear Sky Science · de
Genomisches Sprachmodell reduziert Chimära-Artefakte bei Nanopore-Direct-RNA-Sequencing
Warum das Bereinigen von RNA-Reads wichtig ist
Unsere Zellen lesen ständig genetische Anweisungen, die in RNA geschrieben sind, und neue Sequenziertechnologien erlauben Forschern nun, diesen Prozess mit beispielloser Detailtreue zu beobachten. Eines der leistungsfähigsten Werkzeuge, das Nanopore-Direct-RNA-Sequencing, kann komplette RNA-Moleküle in einem Durchgang lesen – doch es führt auch zu Störungen, die so aussehen lassen können, als seien Gene gebrochen und wieder zusammengesetzt, obwohl das in der Realität nie geschieht. Diese Studie stellt DeepChopper vor, ein Softwarewerkzeug, das wie ein Sprachmodell für Genome funktioniert und diese Fehler bereinigt, sodass Forschende den RNA-Daten vertrauen können.
Wenn der Sequencer falsche Genmischungen erfindet
Moderne Nanopore-Geräte ziehen einzelne RNA-Stränge durch winzige Poren und lesen deren Sequenz direkt aus. Das bietet große Vorteile gegenüber älteren Methoden, etwa das Bewahren chemischer Modifikationen und das Erfassen vollständiger Transkripte in einem einzigen Read. Der Prozess nutzt jedoch auch kurze Helferstücke, sogenannte Adapter, die während der Bibliotheksvorbereitung an RNA-Moleküle angeklebt werden. Manchmal werden zwei oder mehr RNA-Moleküle durch diese Adapter versehentlich miteinander verbunden, wodurch Chimären entstehen – hybride Moleküle, die verschiedene Gene zu fusionieren scheinen. Standard-Analysetools können diese technischen Überreste fälschlich als echte biologische Ereignisse deuten, etwa krebsspezifische Genfusionen oder ungewöhnliche Spleißmuster, und so zu irreführenden Ergebnissen führen.
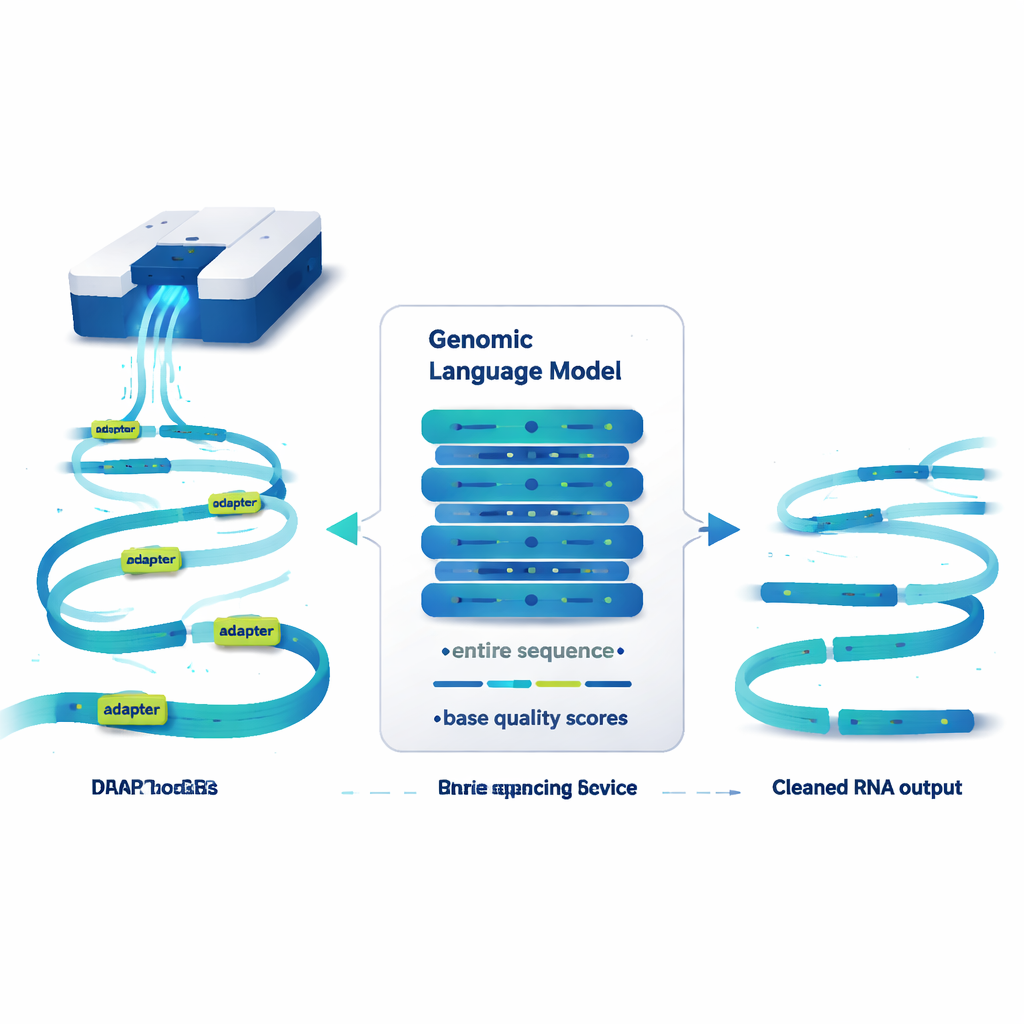
Ein Sprachmodell, das Genome liest, nicht Sätze
DeepChopper behandelt genetische Sequenzen ähnlich wie Text und überträgt Konzepte aus großen Sprachmodellen auf sie. Statt mit Wörtern arbeitet es Buchstabe für Buchstabe und nutzt zusätzlich eine Qualitätsbewertung für jeden Buchstaben, die angibt, wie zuverlässig der Read ist. Aufgebaut auf einer kompakten Architektur namens HyenaDNA kann es bis zu 32.000 Basen gleichzeitig scannen – lang genug, um im Grunde jedes humane RNA-Molekül abzudecken. Für jede einzelne Position schätzt DeepChopper, ob diese Base Teil einer echten RNA-Sequenz oder Teil eines Adapters ist. Ein Verfeinerungsschritt glättet diese Vorhersagen anschließend, sodass Adapter als zusammenhängende Blöcke markiert werden statt als verstreute Punkte.
Die schlechten Verbindungen entfernen, ohne Daten zu vernichten
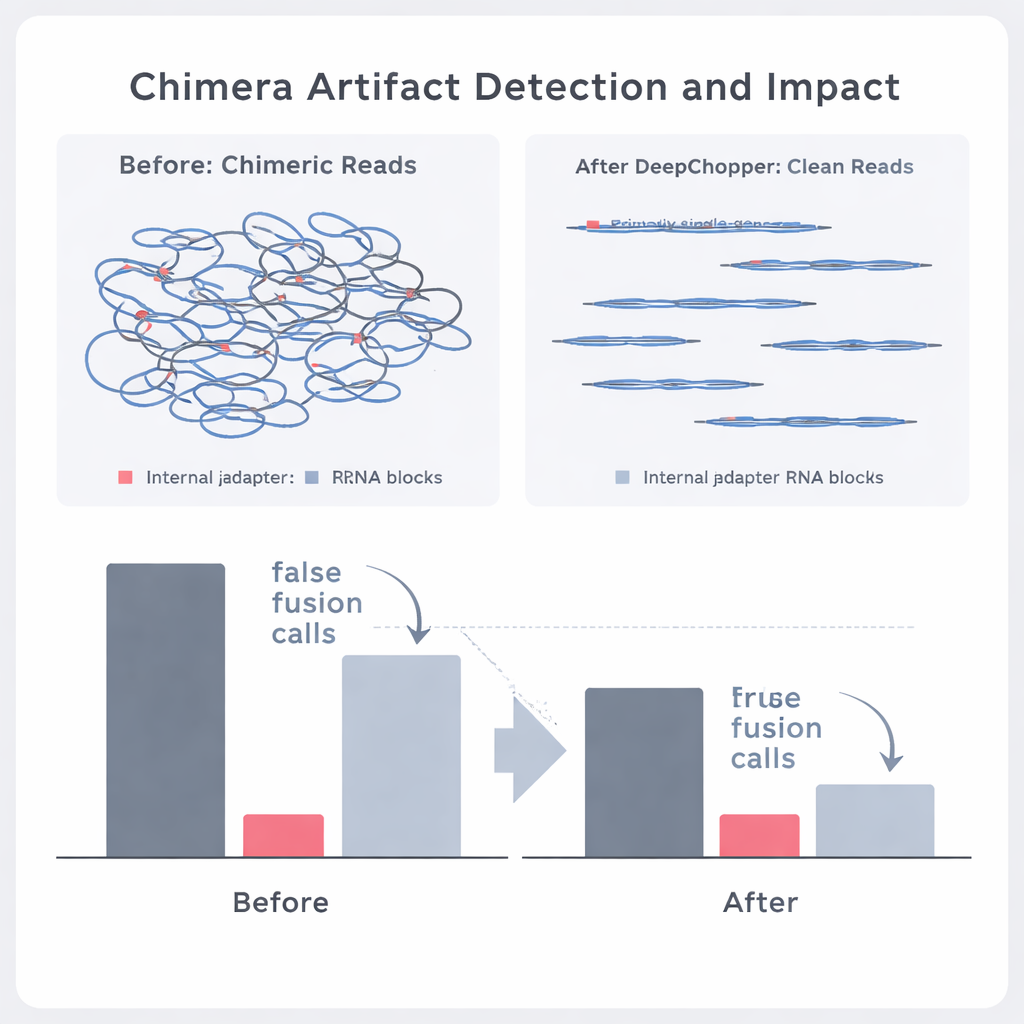
Wenn DeepChopper Adapter innerhalb eines Reads identifiziert hat, macht es etwas Entscheidendes: Anstatt den gesamten Read zu verwerfen, „zerschneidet“ es an den Adapterstellen und bewahrt die echten Abschnitte. Auf diese Weise kann eine künstliche Fusion von zwei RNAs wieder in ihre ursprünglichen Teile zerlegt werden. In Tests mit Millionen von Nanopore-Reads aus mehreren humanen Krebszelllinien und Stammzellen übertraf DeepChopper deutlich bestehende Adapter-Trimming-Tools, die nie für das Direct-RNA-Setting ausgelegt waren. Es erkannte Adapter in synthetischen Benchmarks mit über 99 % Präzision und Recall und skaliert effizient auf Datensätze mit mehr als 20 Millionen Reads unter Einsatz von Grafikprozessoren.
Echte Genfusionen von Sequenzier-Spiegelbildern trennen
Die Autorinnen und Autoren fragten dann, ob DeepChopper echte biologische Ereignisse von Artefakten in realen Krebsdaten unterscheiden kann. Durch den Vergleich der Direct-RNA-Reads mit passenden Datensätzen, die mit unabhängigen Methoden erzeugt wurden (z. B. direct cDNA-Sequencing auf Oxford Nanopore- und PacBio-Plattformen), konnten sie festhalten, welche scheinbaren Chimären von anderen Technologien gestützt wurden und welche nicht. DeepChopper verringerte nicht unterstützte chimerische Alignments um 62–91 % und reicherte gleichzeitig den Anteil der durch andere Methoden bestätigten Fälle stark an. Außerdem reduzierte es die Anzahl verdächtiger Genfusionsaufrufe um fast 90 %, insbesondere solche, die ribosomale Gene betrafen und sich als häufige Artefakte entpuppten. Gleichzeitig blieben echte Fusionsereignisse, die durch Short-Read-RNA-Sequenzierung gestützt wurden, erhalten.
Bessere Chemie hilft – aber Artefakte bleiben
Oxford Nanopore hat kürzlich ein aktualisiertes Sequenzierkit (RNA004) veröffentlicht, das teilweise dazu entwickelt wurde, technische Artefakte zu reduzieren. DeepChopper wurde „out of the box“ auf Daten dieser neuen Chemie angewendet und fand dennoch, dass ein kleiner, aber wichtiger Anteil der Reads interne Adapter und chimerische Verbindungen enthielt. Selbst ohne zusätzliches Training reduzierte das Modell artefaktische Chimären um etwa ein Fünftel; nach Feintuning auf die neuen Daten verbesserte sich die Leistung leicht, ohne echte Signale zu beeinträchtigen. Über alle Chemien und Zelltypen hinweg führte die Korrektur dieser Artefakte dazu, dass nachgelagerte Werkzeuge viele mehr vollständige und alternative Transkripte erkennen konnten, was ein klareres Bild der RNA-Landschaft der Zelle lieferte.
Was das für künftige RNA-Studien bedeutet
Für Nicht-Spezialisten lautet die Kernbotschaft: Nicht jede überraschende RNA-Verbindung, die ein Sequencer meldet, ist echte Biologie – einige sind elektrische Fehler, die durch die Technologie selbst eingeführt werden. DeepChopper wirkt wie ein hochqualifizierter Korrektor für Nanopore-RNA-Daten, erkennt die typischen Adaptersequenzen, die unzusammenhängende Moleküle verbinden, und schneidet sie mit Einzel-Basen-Präzision heraus. Das Ergebnis sind sauberere, verlässlichere Karten darüber, welche RNA-Moleküle in einer Zelle existieren und wie sie zusammengesetzt sind. Da Labore verstärkt auf Long-Read-RNA-Sequenzierung setzen, um Krebs, Hirnerkrankungen und andere komplexe Krankheiten zu untersuchen, werden Werkzeuge wie DeepChopper entscheidend, um rauschende Rohdaten in verlässliche biologische Erkenntnisse zu verwandeln.
Zitation: Li, Y., Wang, TY., Guo, Q. et al. Genomic language model mitigates chimera artifacts in nanopore direct RNA sequencing. Nat Commun 17, 1864 (2026). https://doi.org/10.1038/s41467-026-68571-5
Schlüsselwörter: Nanopore-RNA-Sequenzierung, chimerische Reads, Artefakte von Genfusionen, genomisches Sprachmodell, DeepChopper