Clear Sky Science · de
Endotheliales FUNDC1 reguliert die metabolische Umprogrammierung und den Übergang von Fettleibigkeit zu Diabetes über die SIRT3/GATA2/Endothelin‑1‑Achse
Warum Zellen der Blutgefäße bei Fettleibigkeit und Diabetes eine Rolle spielen
Fettleibigkeit und Typ‑2‑Diabetes werden oft dem Fettgewebe, der Leber oder der Bauchspeicheldrüse zugeschrieben. Diese Studie zeigt jedoch einen überraschenden Mitspieler: die dünne Zellschicht, die unsere Blutgefäße auskleidet — die Endothelzellen. Die Forschenden zeigen, dass ein kleines mitochondriales Protein in diesen Zellen, FUNDC1 genannt, den Körper von einfachem Gewichtszuwachs in einen ausgewachsenen Diabetes treiben kann, indem es beeinflusst, wie die Gefäße mit metabolischen Organen kommunizieren.
Stress auf der inneren „Haut“ des Körpers
Endothelzellen bilden ein weites, sensibles Netzwerk, das den Blutfluss, die Nährstoffzufuhr und Signale an umliegendes Gewebe steuert. Unter gesunden Bedingungen halten sie entspannende Faktoren wie Stickstoffmonoxid und verengende Faktoren wie Endothelin‑1 (ET‑1) im Gleichgewicht. Bei Fettleibigkeit und frühem Diabetes verschiebt sich dieses Gleichgewicht zugunsten von ET‑1, das nicht nur Blutgefäße verengt, sondern auch stört, wie Fett-, Muskel‑ und Leberzellen Zucker und Fette verarbeiten. Die Autorinnen und Autoren zeigen zunächst an Mäusen, dass Gefäßdysfunktion bereits nach nur zwei Monaten einer fettreichen Ernährung auftritt — noch bevor eindeutige Insulinresistenz entsteht — was nahelegt, dass geschädigtes Endothel zur Auslösung metabolischer Erkrankungen beitragen kann, statt nur darauf zu reagieren.
Ein mitochondrialer Schalter, der Körperfett formt
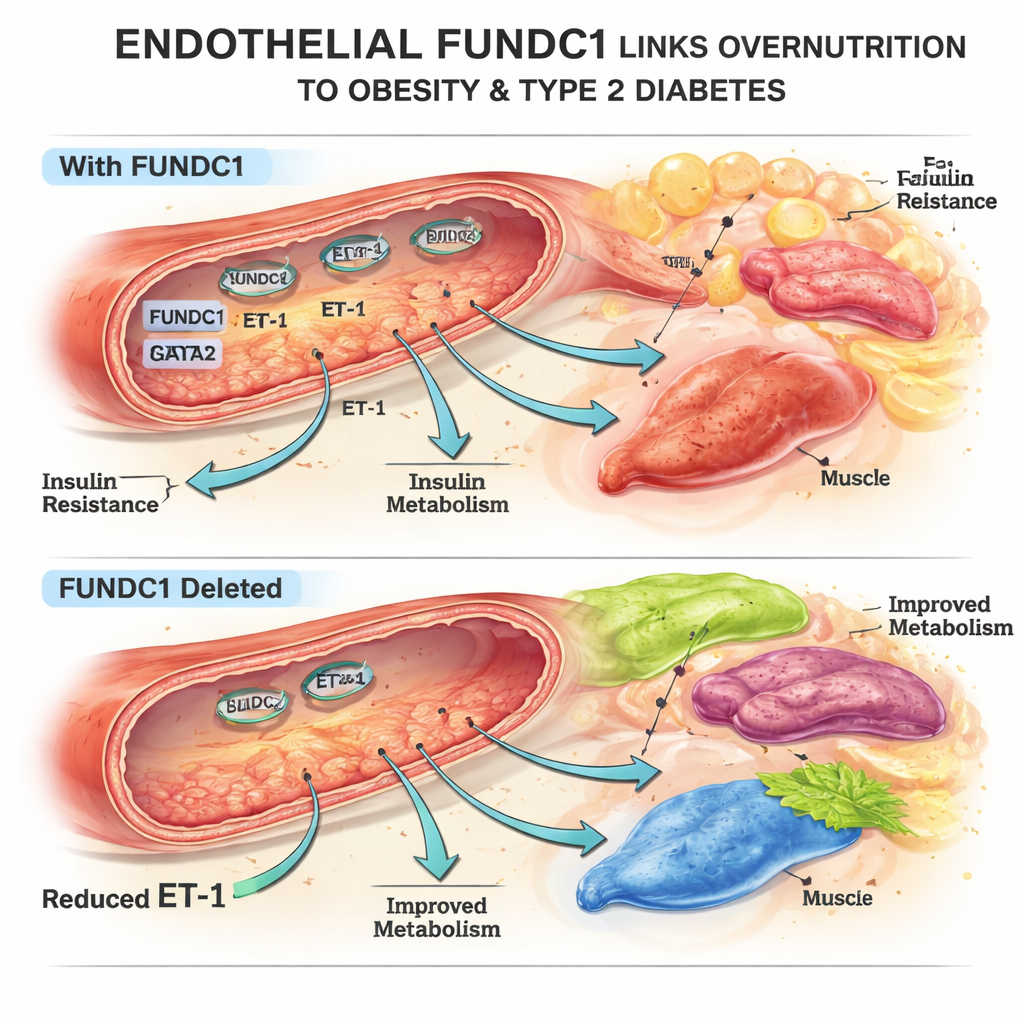
Das Team konzentrierte sich auf FUNDC1, ein Protein auf der Oberfläche von Mitochondrien, den Energie‑Fabriken der Zelle. In Mäusen und menschlichen Endothelzellen, die überschüssigem Fett ausgesetzt waren, änderten sich die FUNDC1‑Spiegel im Zeitverlauf: zunächst sanken sie, bei länger andauernder Überernährung stiegen sie dann stark an. Mit genetisch veränderten Mäusen, denen FUNDC1 nur in Endothelzellen fehlte, fanden die Forschenden heraus, dass diese Tiere teilweise vor einer Gewichtszunahme durch fettreiche Ernährung geschützt waren: Sie hatten weniger Körperfett, kleinere Fettzellen und eine bessere Blutzuckerregulation. Fett‑, Leber‑ und braunes‑Fett‑Gewebe reagierten stärker auf Insulin, obwohl die Insulinspiegel selbst nicht erhöht waren. Diese Veränderungen ließen sich nicht durch Unterschiede bei Nahrungsaufnahme oder Aktivität erklären, sondern deuten darauf hin, dass das Gefäßsystem den Stoffwechsel beeinflusst.
Ein chemischer Botenstoff, der Insulinresistenz antreibt
Um herauszufinden, wie endotheliales FUNDC1 entfernte Organe beeinflusst, screente das Team verschiedene von Endothelzellen sezernierte Substanzen. Eine stach hervor: ET‑1. Wenn FUNDC1 in Endothelzellen gelöscht wurde, fiel die ET‑1‑Produktion in den Gefäßen und im Blut deutlich ab — sowohl unter normalen als auch unter fettreichen Bedingungen. Experimente in kultivierten Fett‑, Leber‑ und Muskelzellen zeigten, dass ET‑1 das Wachstum von Vorläufer‑Fettzellen fördert, Speichern und Abbau von Fett verändert und die Fetteinlagerung in Leber und Muskel bei hoher Fettbelastung verschlechtert — ein Muster, das bekanntlich Insulinresistenz begünstigt. Bei lebenden Mäusen zerstörte die Infusion von ET‑1 zu Beginn einer fettreichen Ernährung den Schutz, den das Fehlen von endothelialem FUNDC1 bot: Körpergewicht, Fettmasse, Blutzuckerregulation und Gefäßfunktion verschlechterten sich, was ET‑1 als wichtigen Verbindungspunkt zwischen Endothel und metabolischer Erkrankung unterstreicht.
Eine interne Signalkette: FUNDC1, SIRT3 und GATA2
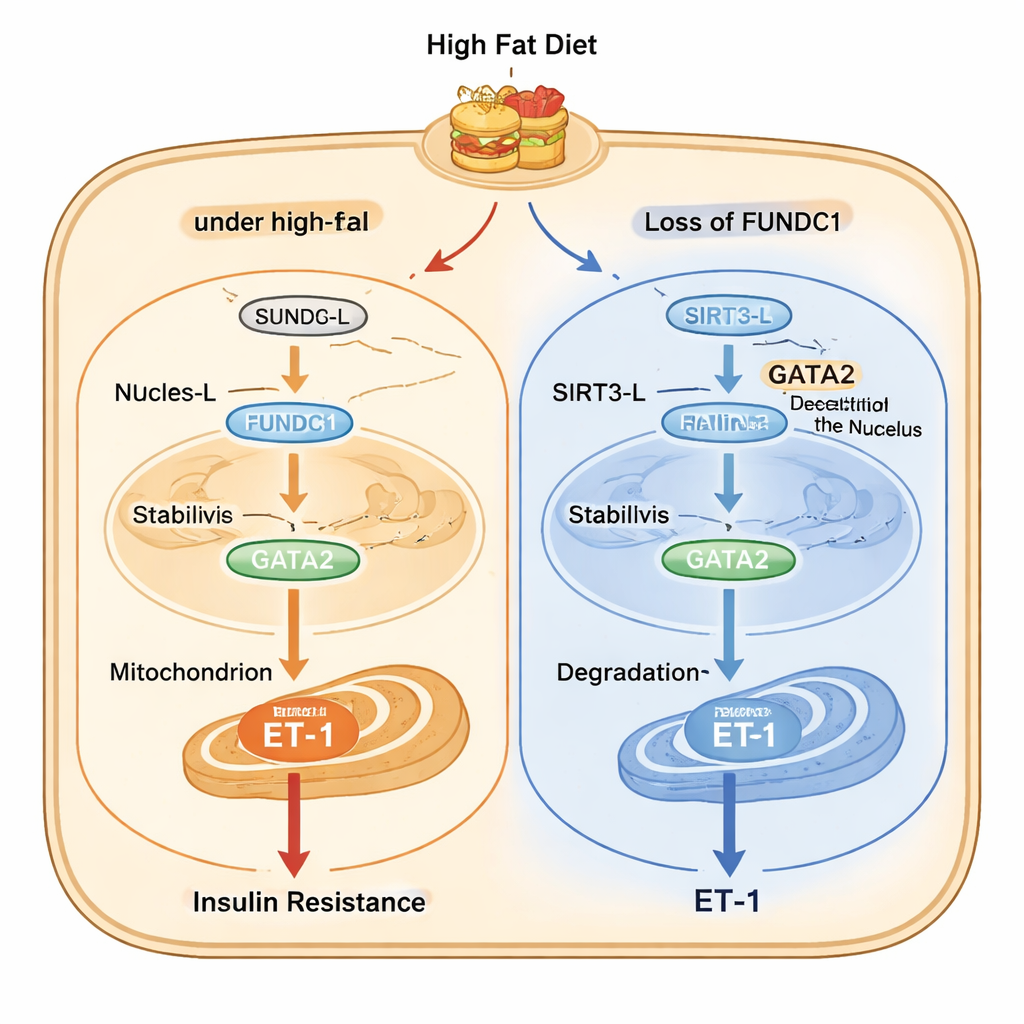
Die Studie beschreibt dann eine detaillierte molekulare Kette innerhalb von Endothelzellen. Unter Fettstress bewegt sich eine lange Form des Enzyms SIRT3 (SIRT3‑L), die sowohl im Nukleus als auch in Mitochondrien vorkommen kann, mit Hilfe von FUNDC1 und einem Chaperon‑Protein namens HSC70 aus dem Zellkern in die Mitochondrien. Einmal in den Mitochondrien gebunden, steht im Nukleus weniger SIRT3‑L zur Verfügung, um Acetylgruppen von GATA2 zu entfernen — einem Transkriptionsfaktor, der die ET‑1‑Genaktivität steigert. Acetyliertes GATA2 ist stabiler und fördert die ET‑1‑Produktion. Fehlt FUNDC1, verbleibt SIRT3‑L im Nukleus, deacetyliert dort GATA2, was zum Abbau von GATA2 und zu geringerer ET‑1‑Produktion führt. Interessanterweise fördert SIRT3 wiederum den Abbau von FUNDC1, wodurch eine Rückkopplung entsteht, die den Weg normalerweise bremst, bei chronischer Überernährung jedoch fehlreguliert wird.
Von Mausmodellen zur menschlichen Erkrankung
Um zu prüfen, ob dieser Mechanismus beim Menschen relevant ist, untersuchten die Forschenden Blut und kleine Arterien von Personen mit Fettleibigkeit und Typ‑2‑Diabetes sowie von gesunden Freiwilligen. Patientinnen und Patienten mit beiden Erkrankungen hatten höhere Blutspiegel von ET‑1 sowie eine höhere Expression von FUNDC1, GATA2 und dem ET‑1‑Gen in ihrem Gefäßendothel. Die ET‑1‑Konzentration im Blut korrelierte eng mit Body‑Mass‑Index und langfristigem Blutzucker (HbA1c), und die Gefäß‑ET‑1‑Genlevel standen in starker Korrelation zu FUNDC1 und GATA2. Diese Muster spiegeln die Ergebnisse bei Mäusen wider und stützen die Idee, dass eine überaktive FUNDC1–SIRT3–GATA2–ET‑1‑Achse in menschlichem Gefäßgewebe unter metabolischem Stress wirksam ist.
Ein neues Ziel im Kampf gegen Diabetes
Für Nichtfachleute ist die Kernbotschaft: Schäden durch Überernährung können sich zuerst in den Zellen zeigen, die unsere Blutgefäße auskleiden. Dort lenkt ein mitochondriales Protein, FUNDC1, ein regulatorisches Enzym, SIRT3, aus dem Zellkern um, sodass ein anderer Faktor, GATA2, die Produktion von ET‑1 erhöht — ein starkes hormonähnliches Signal, das sowohl Gefäßversteifung als auch Insulinresistenz fördert. Die Blockade dieses Weges — etwa durch Reduktion der endothelialen FUNDC1‑Aktivität oder durch Herunterregulieren von ET‑1 — könnte helfen, den Übergang von Fettleibigkeit zu Diabetes zu verhindern und gleichzeitig die Blutgefäße zu schützen.
Zitation: Li, J., Li, D., Zhao, F. et al. Endothelial FUNDC1 regulates metabolic reprogramming and the obesity-diabetes transition through the SIRT3/GATA2/endothelin-1 axis. Nat Commun 17, 1836 (2026). https://doi.org/10.1038/s41467-026-68548-4
Schlüsselwörter: Endothelzellen, Mitochondrien, Endothelin‑1, Fettleibigkeit, Typ‑2‑Diabetes