Clear Sky Science · de
TET1 als Masterregulator, der GPX4‑abhängige und -unabhängige Ferroptose‑Überwachung bei akutem myeloischem Leukämie steuert
Warum diese Forschung für die Krebstherapie wichtig ist
Viele neue Krebsmedikamente zielen darauf ab, bösartige Zellen in einen Selbstzerstörungsmodus namens Ferroptose zu treiben — eine Form des Zelltods, die von Eisen und Lipidschäden getrieben wird. Dennoch sind einige Tumoren hartnäckig resistent gegen diesen Ansatz. Diese Studie zeigt, wie ein DNA‑modifizierendes Protein namens TET1 Leukämiezellen hilft, der Ferroptose durch zwei unterschiedliche biochemische Abwehrsysteme zu entgehen — und demonstriert, dass das Blockieren dieser Abwehrmechanismen selbst resistente Tumoren verwundbar machen kann.
Eine tödliche Mischung aus Eisen und beschädigten Fetten
Ferroptose tritt auf, wenn Eisen die unkontrollierte Oxidation von Lipiden in Zellmembranen antreibt, was schließlich zum Platzen der Zellen führt. Bei akuter myeloischer Leukämie (AML), wie bei vielen Krebsarten, setzen Zellen starke Überwachungssysteme ein, um diesen Prozess in Schach zu halten. Ein zentraler Wächter ist das Enzym GPX4, das ein kleines Molekül namens Glutathion nutzt, um schädliche Lipidperoxide zu neutralisieren. Weitere Reserve‑Systeme erzeugen Antioxidantien, die gefährliche Radikale abfangen können, selbst wenn GPX4 beeinträchtigt ist. Zu wissen, welche Master‑Schalter diese Abwehr koordinieren, ist entscheidend für die Entwicklung von Therapien, die zuverlässig Ferroptose in Krebszellen auslösen, während gesundes Gewebe geschont wird.
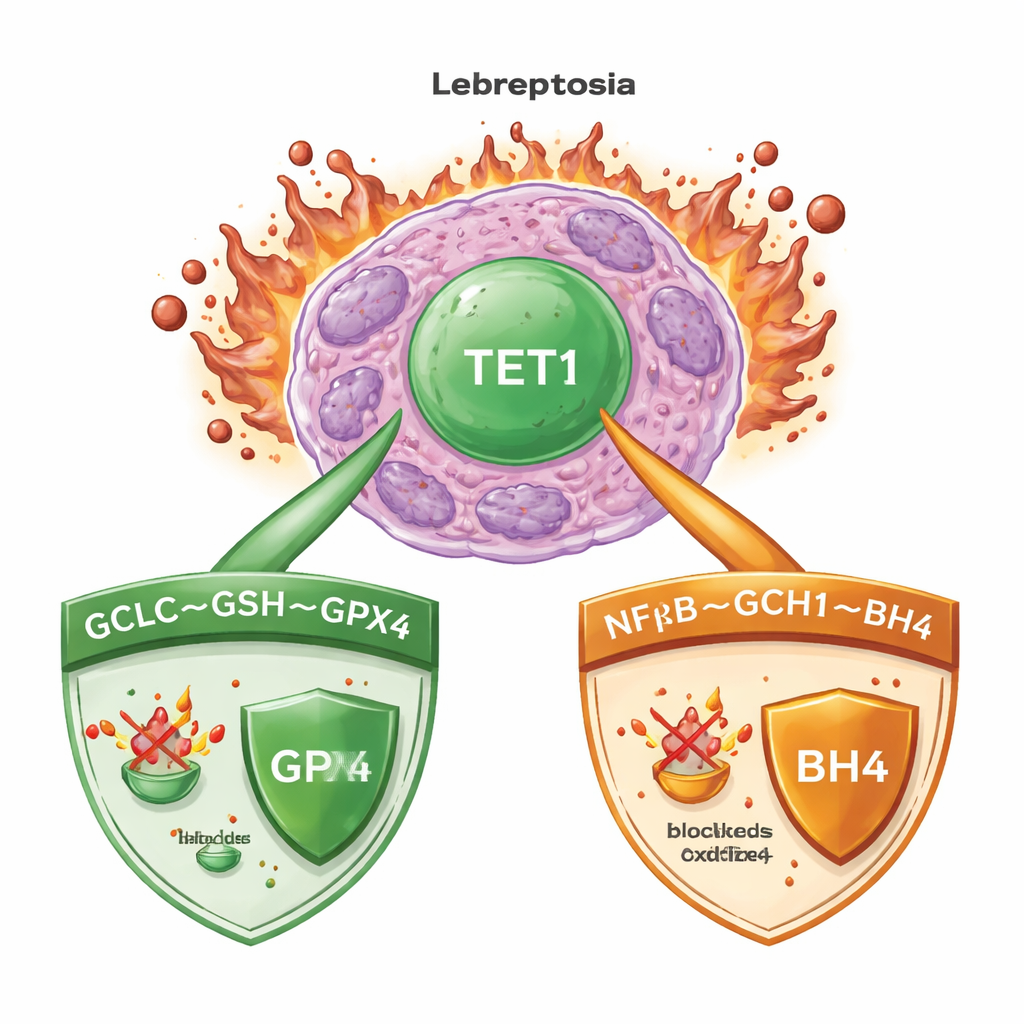
TET1 tritt als zentrales Steuerzentrum hervor
Die Forscher verglichen Dutzende von Krebszellproben, darunter viele AML‑Linien und patientenabgeleitete Zellen, und beobachteten ein klares Muster: Zellen, die Ferroptose widerstanden, wiesen höhere TET1‑Spiegel auf — ein Enzym, das chemische DNA‑Marken verändert und so die Genaktivität beeinflusst. Reduzierten sie TET1 mit genetischen Werkzeugen oder hemmten seine Aktivität mit einem niedermolekularen Wirkstoff, wurden die Krebszellen deutlich empfänglicher für Ferroptose‑induzierende Substanzen. Dies galt sowohl in Zellkulturen als auch in Mausmodellen der AML. Umgekehrt schützte eine erhöhte TET1‑Expression Zellen vor ferroptotischem Tod und verringerte die Ansammlung reaktiver Sauerstoffspezies, die chemisch aggressiven Nebenprodukte, die Membranschäden vorantreiben.
Stärkung des wichtigsten Antioxidansschilds
Bei genauerer Betrachtung kartierte das Team, wo TET1 im Genom wirkt, und fand, dass es das Gen GCLC direkt aktiviert. GCLC codiert ein entscheidendes Enzym, das die Produktion von Glutathion anstößt — dem Treibstoff für GPX4. Durch Erhöhung einer spezifischen DNA‑Marke (5‑Hydroxymethylcytosin) im Promotor von GCLC steigert TET1 die Glutathionsynthese. Unter normalen Nährstoffbedingungen vergrößert dies den wichtigsten Antioxidans‑Vorrat der Zelle. Bei Cystinmangel produziert dasselbe Enzymkomplex ungewöhnliche γ‑Glutamyl‑Peptide, die helfen, überschüssiges Glutamat zu binden — ein weiterer Weg, Ferroptose abzuschwächen. Sowohl in kultivierten Zellen als auch in Mäusen führten der Verlust von TET1 oder die pharmakologische Hemmung der Glutathionsynthese zu stark erniedrigten Glutathionwerten und geringeren schützenden Peptiden, wodurch Leukämiezellen deutlich anfälliger für Ferroptose‑Auslöser wurden.
Ein zweiter, GPX4‑unabhängiger Fluchtweg
Überraschenderweise endete TET1s Schutzfunktion nicht mit der Glutathion‑GPX4‑Achse. Selbst wenn GPX4 aus Leukämiezellen entfernt wurde, konnte zusätzliches TET1 weiterhin ferroptotischen Tod verhindern, was auf eine zweite Verteidigungslinie hindeutet. Die Autoren führten dies auf die Aktivierung des NFκB‑Signalwegs durch TET1 zurück, insbesondere auf eine Komponente namens NFKB2. Diese wiederum erhöht die Expression von GCH1, einem Enzym, das das Antioxidans BH4 produziert. BH4 kann Membranlipide vor Oxidation schützen, ohne auf GPX4 angewiesen zu sein. Wurde GCH1 genetisch ausgeschaltet oder chemisch blockiert, ging die Schutzwirkung von TET1 gegen Ferroptose teilweise verloren. Zusammen definieren diese Befunde eine TET1–NFKB2–GCH1‑Achse, die ein GPX4‑unabhängiges Ferroptose‑Überwachungssystem bildet.
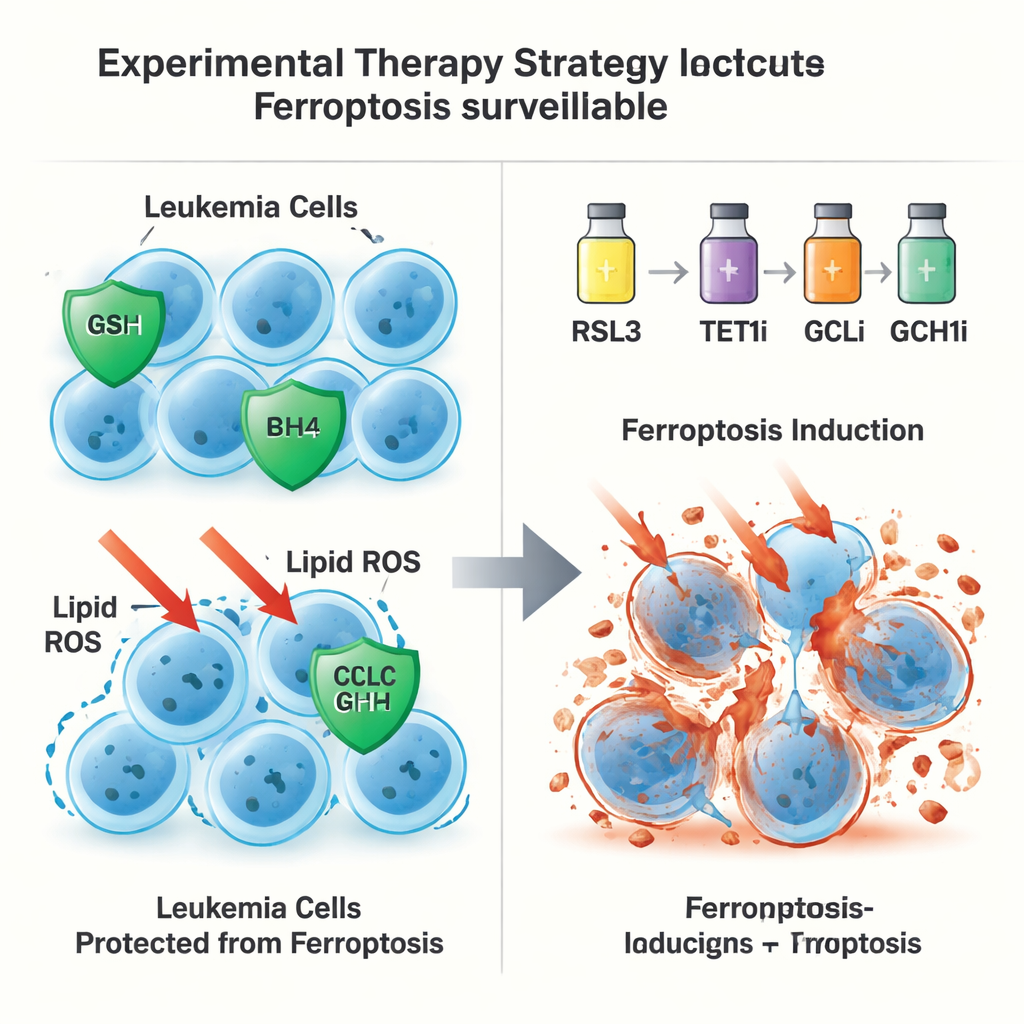
Eine Schwäche in eine therapeutische Chance verwandeln
Mit dieser Karte der Doppelwege testeten die Forscher, ob das gleichzeitige Anstoßen von Ferroptose und das Schwächen der TET1‑kontrollierten Abwehr einen therapeutischen Vorteil bringen könnte. In Maus‑AML‑Modellen und patientenabgeleiteten Leukämie‑Transplantaten in Mäusen reduzierten niedrige Dosen eines Ferroptose‑induzierenden Wirkstoffs kombiniert mit Inhibitoren von TET1, der GSH‑Synthese (über GCLC) oder GCH1 die Leukämielast drastisch, verlängerten das Überleben und dezimierten Leukämieinitiierende Zellpopulationen. Wichtig ist, dass der Ferroptose‑Induktor in einer Bruchteil der Dosen eingesetzt wurde, die in früheren Studien berichtet wurden, wodurch Bedenken hinsichtlich der Toxizität für normale blutbildende Stammzellen verringert wurden.
Was das für zukünftige Krebstherapien bedeutet
Für Nicht‑Spezialisten lautet die Kernbotschaft: Leukämiezellen überleben durch den Betrieb zweier sich überschneidender antioxidativer „Schilde“, die beide von TET1 koordiniert werden — einer fokussiert auf Glutathion und GPX4, der andere auf GCH1 und BH4. Die Arbeit zeigt, dass durch moderate Aktivierung der Ferroptose bei gleichzeitigem Blockieren von TET1 und seinen nachgeschalteten Partnern Ärzte eines Tages möglicherweise Resistenzen überwinden und Krebszellen selektiv an den Rand bringen können, ohne gesundes Gewebe zu überwältigen. Obwohl diese Strategien noch nicht klinisch anwendbar sind, identifiziert die Studie TET1 als einen mächtigen Kontrollknoten und vielversprechendes Ziel für Kombinationstherapien bei AML und potenziell anderen schwer behandelbaren Tumoren.
Zitation: Yang, L., Lu, J., Yun, W. et al. TET1 as a master regulator controlling GPX4-dependent and -independent ferroptosis surveillance in acute myeloid leukemia. Nat Commun 17, 1800 (2026). https://doi.org/10.1038/s41467-026-68509-x
Schlüsselwörter: Ferroptose, akute myeloische Leukämie, TET1, Glutathion, Krebs‑Epigenetik