Clear Sky Science · de
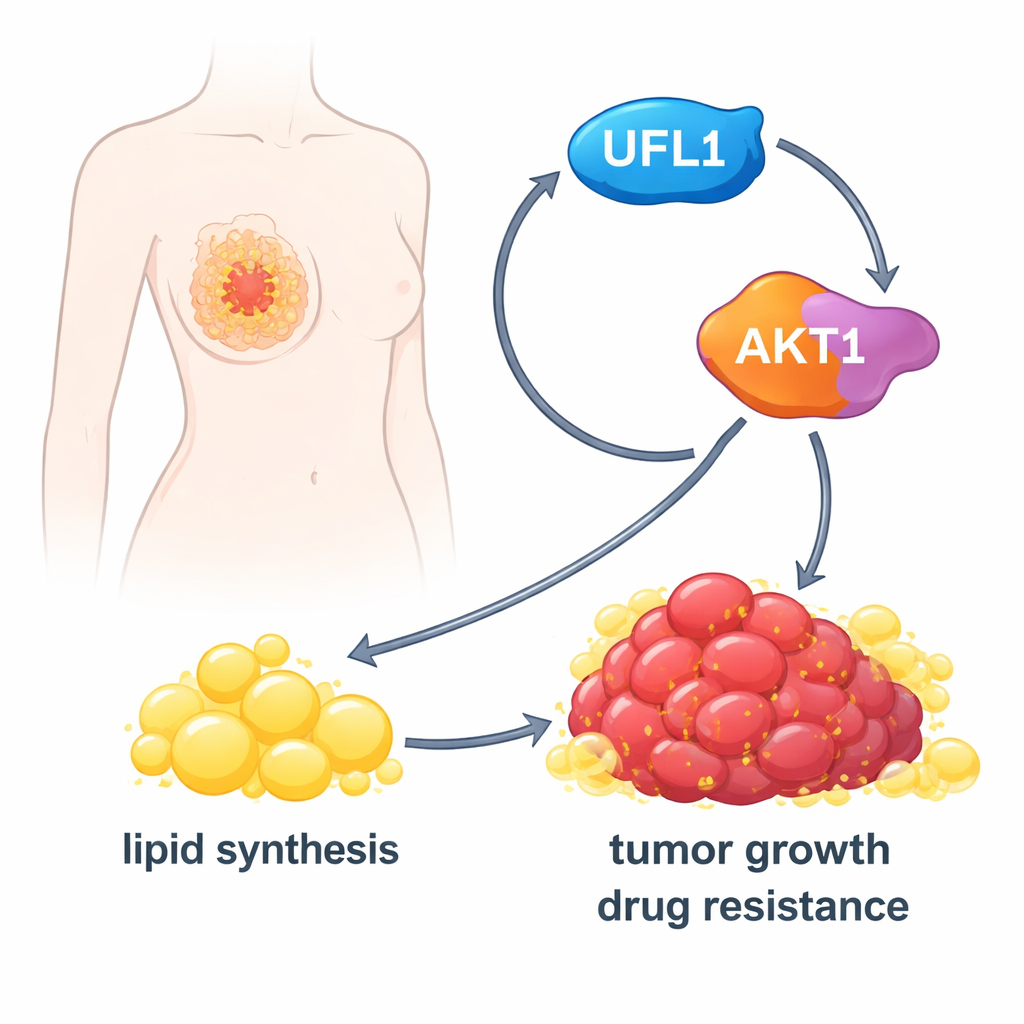
Die UFL1‑AKT-Positive‑Feedback‑Schleife fördert das Fortschreiten von Brustkrebs durch Verstärkung der Lipidsynthese
Warum Krebszellen so sehr auf Fett angewiesen sind
Brustkrebszellen brauchen wie alle schnellwachsenden Zellen eine konstante Versorgung mit Bausteinen, um neue Membranen und Signalstoffe zu bilden. Fette beziehungsweise Lipide sind ein zentraler Teil dieser Versorgung. Diese Studie zeigt, wie ein bislang wenig bekanntes Proteinsystem Krebszellen in einen „Fettproduktions‑Übermodus“ versetzt, der das Tumorwachstum fördert und Therapien erschwert. Das Verständnis dieser versteckten Treibstoffquelle könnte neue Wege eröffnen, Brustkrebserkrankungen zu verlangsamen oder auszuhungern.
Ein verstecktes Protein‑Tag mit großen Folgen
Unsere Zellen regulieren das Verhalten ihrer Proteine ständig mit winzigen chemischen Anhängen. Ein solcher Anhängsel, genannt UFM1, wird von einem Enzym namens UFL1 angehängt. Die UFM1‑Markierung (UFMylierung) wurde mit DNA‑Reparatur und Stressantworten in Verbindung gebracht, ihre Rolle beim Krebs war aber unklar. Die Autorinnen und Autoren zeigen, dass UFL1 in menschlichen Brusttumoren deutlich aktiver ist als im normalen Brustgewebe, über alle Hauptsubtypen hinweg. Patientinnen deren Tumoren höhere UFL1‑Spiegel aufweisen, haben tendenziell eine schlechtere Überlebensprognose. Reduzierte man UFL1 in Brustkrebszellen oder in Maus‑Tumoren, verlangsamte sich das Krebswachstum, die Zellteilung nahm ab und die Zellsterblichkeit stieg — Hinweise darauf, dass UFL1 wie ein tumorfördernder Faktor wirkt.
Wie ein Signalzentrum in den Fettmodus geschaltet wird
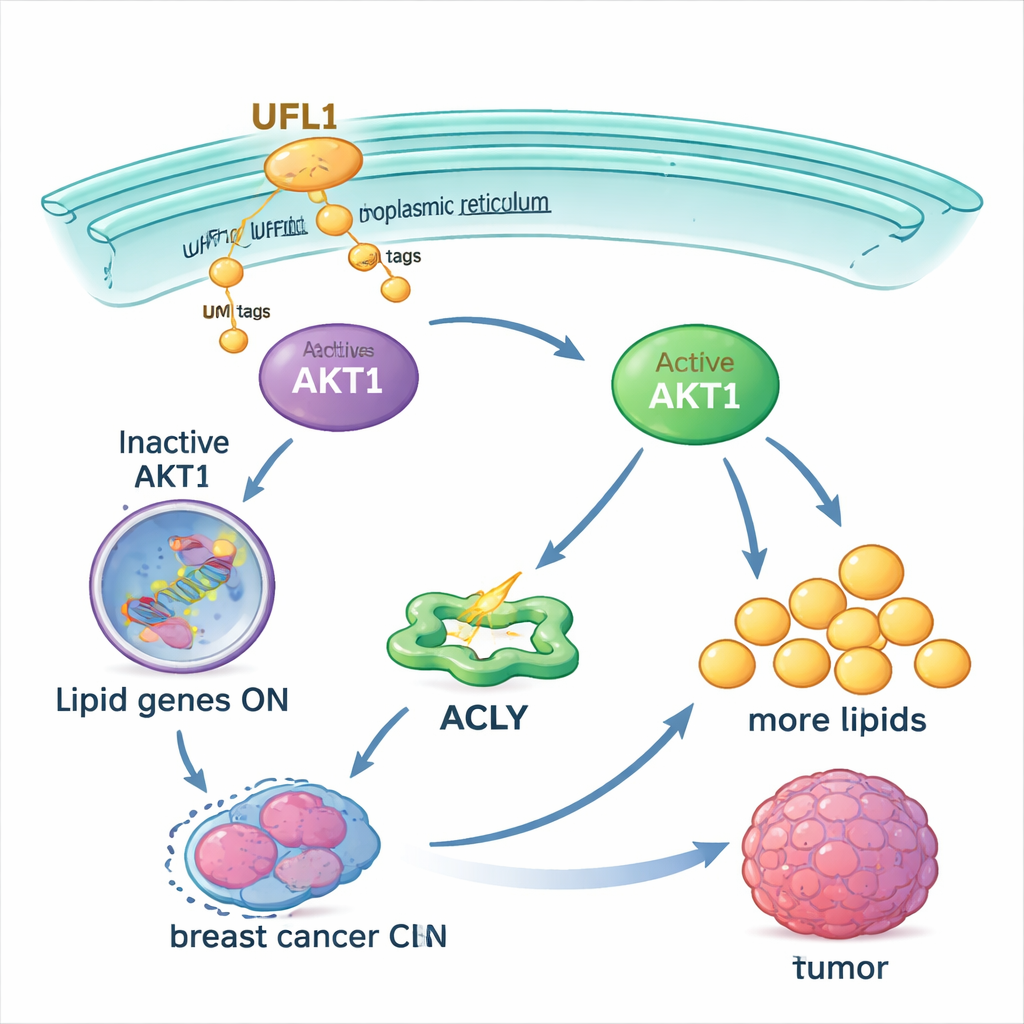
Um zu verstehen, wie UFL1 Tumoren unterstützt, suchte das Team nach seinen molekularen Partnern und fand ein zentrales Signalkontrollprotein namens AKT1. AKT1 ist ein zentraler Schalter, der Zellen zum Wachsen und zur Lipidsynthese anregt. Die Studie zeigt, dass UFL1 physisch an AKT1 bindet und es an mehreren spezifischen Stellen mit UFM1 markiert. Diese Markierung verändert die Form von AKT1, lockert eine interne „Bremse“, die es normalerweise inaktiv hält, und unterstützt die Verlagerung von AKT1 zu einer Membrankompartimentierung im Inneren der Zelle, dem endoplasmatischen Retikulum, wo andere Enzyme es durch Phosphorylierung vollständig aktivieren können. Ohne UFM1‑Marken an diesen Stellen lässt sich AKT1 deutlich schwerer aktivieren.
Die Krebs‑Fettfabrik einschalten
Einmal aktiviert, treibt AKT1 die Zellen an, ihre Fettproduktion hochzufahren. Es steigert die Aktivität eines wichtigen Stoffwechselenzyms, ACLY, und erhöht die Spiegel von Masterregulatoren wie SREBP1, die viele fettbildende Gene einschalten. In im Labor kultivierten Brustkrebszellen führten UFL1‑Aktivität zu einer Anhäufung von Fetttropfen, freien Fettsäuren und Cholesterin. Das Entfernen von UFL1 verringerte diese Lipidspeicher deutlich, sowohl in Zellkulturen als auch in Tumoren, die in Mäusen wuchsen. Das Zuführen zusätzlicher Fettsäuren von außen konnte das Wachstum von UFL1‑defizienten Krebszellen weitgehend wiederherstellen, was darauf hindeutet, dass die Hauptfunktion von UFL1 darin besteht, genügend Lipide bereitzustellen, um das Tumorwachstum zu stützen.
Eine sich selbst verstärkende Schleife, die Tumorwachstum antreibt
Überraschenderweise verläuft die Beziehung zwischen UFL1 und AKT1 in beide Richtungen. Die Forschenden entdeckten, dass aktiviertes AKT1 seinerseits eine Phosphatmarke an UFL1 an einer bestimmten Stelle anbringt. Diese Modifikation verstärkt wiederum UFL1s Fähigkeit, AKT1 mit UFM1 zu markieren, wodurch eine positive Rückkopplungsschleife entsteht: UFL1 aktiviert AKT1 und AKT1 aktiviert UFL1. Mutationen entweder an den UFM1‑Markierungsstellen von AKT1 oder an der Phosphatstelle von UFL1 zerstören diese Schleife. In Mäusen wuchsen Tumoren mit diesen mutierten Versionen schlecht, enthielten weniger Lipide und zeigten mehr sterbende Zellen. In Proben von Patientinnen mit triple negativem Brustkrebs traten höhere Level von aktiviertem UFL1 und aktiviertem AKT1 häufig gemeinsam auf, was unterstreicht, dass diese Schleife auch in realen Tumoren aktiv ist.
Die Schleife schwächen, um Therapien zu verbessern
Da viele Arzneimittelversuche bereits auf AKT abzielen, prüften die Autorinnen und Autoren, ob das Blockieren der UFMylierung diese Behandlungen wirksamer machen könnte. In Brustkrebszellen senkten ein kleines Molekül als UFMylierungsinhibitor und ein AKT‑Hemmer jeweils die AKT1‑Aktivität und die Lipidansammlung, die Kombination beider wirkte jedoch deutlich stärker als jede Einzelbehandlung. Die Kombination verlangsamte außerdem das Tumorwachstum und reduzierte den Fettgehalt in Mäusetumoren, ohne dass die Tiere auffälligen Gewichtsverlust zeigten. Das Blockieren der UFMylierung machte Krebszellen zudem empfindlicher gegenüber Standardchemotherapien wie Cisplatin und Etoposid, die oft versagen, wenn AKT stark aktiv ist.
Was das für Patientinnen bedeutet
Für eine nichtfachliche Leserin zeigt die Studie, dass sich bestimmte Brustkrebse eine selbstverstärkende Schleife einbauen, die sowohl ihr Wachstum als auch ihre Fettversorgung antreibt. UFL1 und AKT1 wirken zusammen wie zwei gleichzeitig durchgetretene Pedale: das eine markiert, das andere signalisiert, und gemeinsam treiben sie die Lipidproduktion und das Tumorwachstum an. Durch das Finden von Wirkstoffen, die diese Schleife unterbrechen — sei es durch Hemmung der UFMylierung, von AKT1 oder ihrer Wechselwirkung — könnten künftige Therapien das Tumorwachstum verlangsamen und bestehende Behandlungen wirksamer machen, insbesondere bei aggressiven Brustkrebsarten.
Zitation: Meng, F., Du, Y., Liang, J. et al. The UFL1-AKT positive feedback loop promotes breast cancer progression by enhancing lipid synthesis. Nat Commun 17, 614 (2026). https://doi.org/10.1038/s41467-026-68492-3
Schlüsselwörter: Brustkrebs, Lipidstoffwechsel, AKT-Signalgebung, UFMylierung, Zielgerichtete Therapie