Warum medikamentenresistenter Lungenkrebs wichtig ist
Gezielte Wirkstoffe haben die Behandlung bestimmter Lungenkrebserkrankungen verändert, indem sie ein fehlerhaftes Wachstumssignal namens EGFR ins Visier nehmen. Doch bei den meisten Patientinnen und Patienten hören diese Medikamente nach ein bis zwei Jahren auf zu wirken, weil der Tumor Resistenzen entwickelt. Diese Studie enthüllt eine überraschende Wendung: Sobald Tumoren gegenüber EGFR-Inhibitoren resistent werden, entsteht eine neue Achillesferse, die mit einem anderen Wirkstofftyp angreifbar ist. Das Verständnis dieser verborgenen Schwäche könnte künftige Therapiestrategien inspirieren, die die Evolution des Krebses einkreisen, statt ihr hinterherzujagen.
Eine verborgene Schwäche offenbart
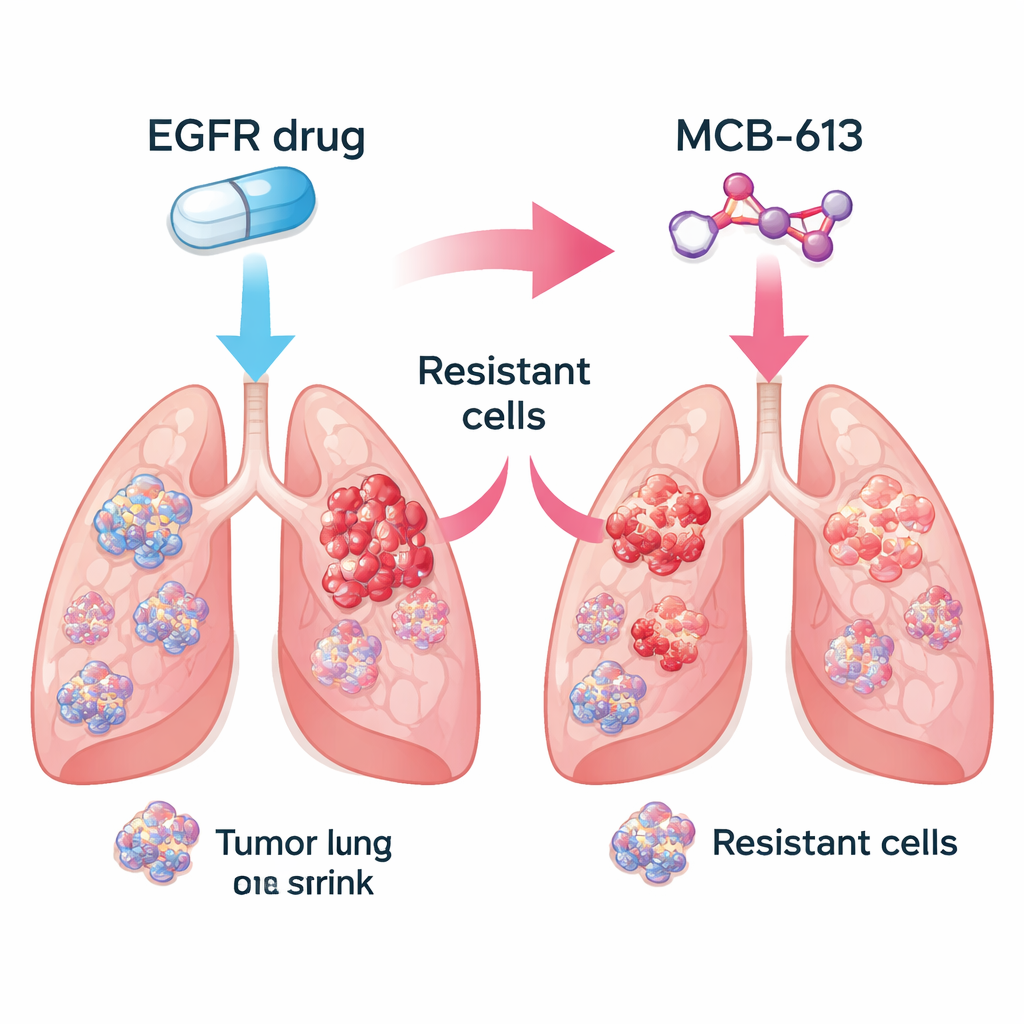
Die Forschenden konzentrierten sich auf nicht-kleinzelligen Lungenkrebs, der durch mutiertes EGFR angetrieben wird – eine verbreitete Form der Erkrankung. Im Labor verglichen sie medikamentenempfindliche Krebszellen mit eng verwandten Zellen, die Resistenzen gegen EGFR-blockierende Medikamente wie Gefitinib und Osimertinib entwickelt hatten. Anschließend testeten sie eine Bibliothek von rund 2.100 kleinen Molekülen, um herauszufinden, welche davon die resistenten Zellen wirksamer abtöten als die ursprünglichen, medikamentensensitiven Zellen. Unter vielen Kandidaten fiel eine Verbindung, MCB-613 genannt, beständig auf. Resistente Zellen, die EGFR-Inhibitoren trotzten, erwiesen sich sowohl in Zellkulturen als auch in Maus-Tumoren als ungewöhnlich verwundbar gegenüber MCB-613.
Gemischte Tumorpopulationen einkreisen Figure 1.
Reale Tumoren bestehen aus Mischungen von Zellen: Einige bleiben gegenüber dem ursprünglichen Medikament empfindlich, während andere durch verschiedene genetische Mechanismen Resistenz erwerben. Das Team fragte, ob die Kombination eines EGFR-Inhibitors mit MCB-613 diese Diversität auslöschen könnte. In einem kontrollierten Experiment mischten sie überwiegend medikamentensensitive Zellen mit einem kleinen Anteil verschiedener resistenter Typen, um einen Patiententumor zu simulieren. Die Behandlung dieser gemischten Population mit entweder dem EGFR-Inhibitor oder MCB-613 allein ließ einige Zellen überleben und wieder wachsen. Wurden jedoch beide Wirkstoffe gleichzeitig eingesetzt, brach die gesamte Population zusammen. Das deutet darauf hin, dass die Kombination einer Standardzieltherapie mit einem gezielt gewählten "kollateralen Sensitivitäts"-Wirkstoff Tumoren in eine evolutionäre Sackgasse treiben könnte.
Eine molekulare Brücke, die einen Wächter zerstört
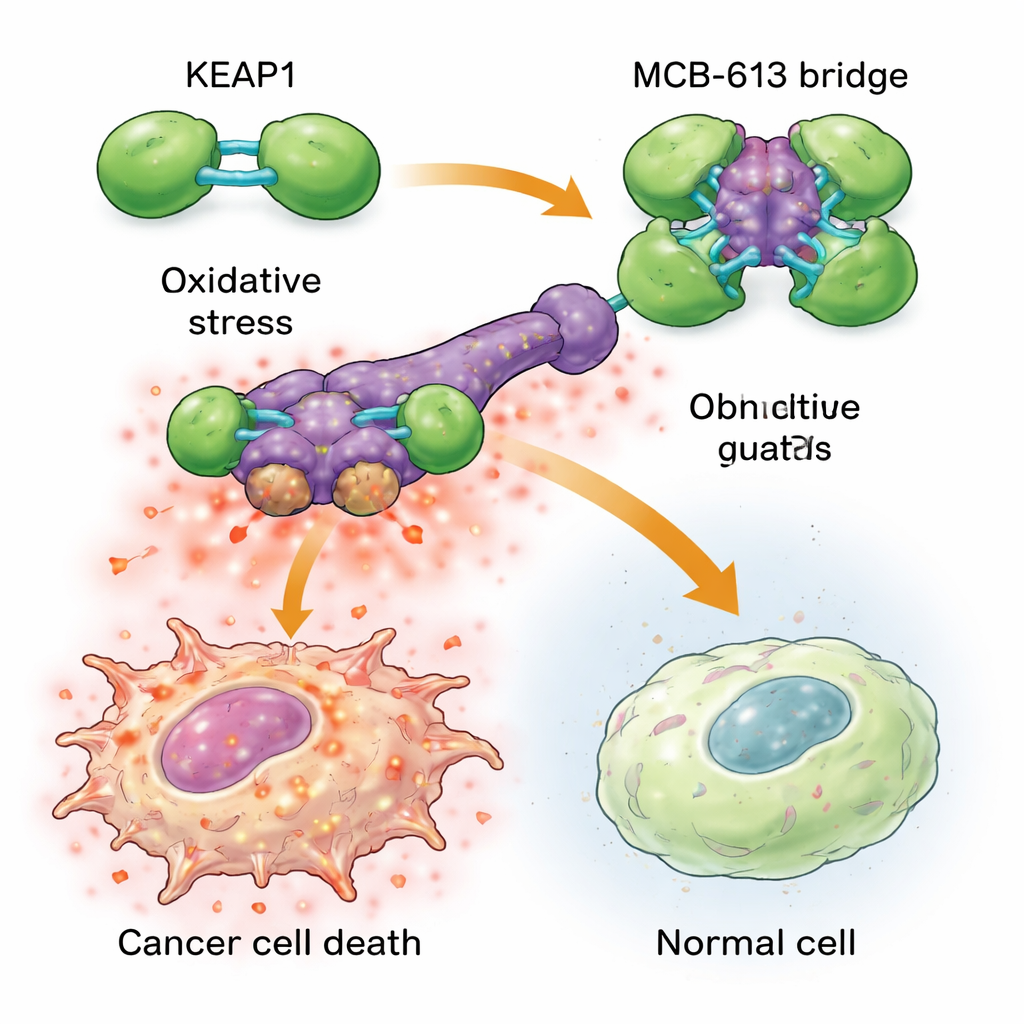
Um zu verstehen, warum MCB-613 resistente Zellen so stark trifft, untersuchten die Forschenden, an welche Proteine die Verbindung bindet. Mithilfe chemischer Sonden und eines gezielten CRISPR-Gen-Screens identifizierten sie ein Protein namens KEAP1 als essentiell für die Wirkung von MCB-613. KEAP1 fungiert normalerweise als zellulärer Wächter, der Stress erkennt und schützende Antworten reguliert. Das Team fand heraus, dass MCB-613 auf ungewöhnliche Weise an KEAP1 andockt: Es wirkt wie eine starre molekulare Brücke, die KEAP1-Einheiten zu übergroßen, abnormalen Clustern verbindet. Dieser Prozess hängt nicht von den üblichen reaktiven Schwefelstellen in KEAP1 ab, sondern von einer spezifischen Lysin-Aminosäure in seinem Dimerisierungsbereich. Wenn dieses Lysin mutiert war, konnte MCB-613 KEAP1 nicht mehr verklumpen und resistente Zellen zeigten keine Überempfindlichkeit gegenüber der Verbindung mehr.
Hilfreichen Stress in tödliche Überlastung verwandeln Figure 2.
Das Verklumpen von KEAP1 löst in medikamentenresistenten Krebszellen eine gefährliche Kettenreaktion aus. Diese Zellen leben bereits unter höherem Grundstress, mit erhöhten Spiegeln reaktiver Sauerstoffspezies (schädliche chemische Nebenprodukte) und erhöhter Aktivität eines Schutznetzwerks, das als integrierte Stressantwort bekannt ist. Wird MCB-613 hinzugefügt, treibt die Störung von KEAP1 diesen belasteten Zustand über die Grenze: Reaktive Sauerstoffverbindungen häufen sich weiter an, und Schlüsselfaktoren der Stressantwort wie ATF4 und CHOP schalten mächtige Todesprogramme ein. Das Blockieren dieser Stressregulatoren oder das chemische Binden reaktiver Sauerstoffspezies schützte die Zellen weitgehend vor MCB-613. Interessanterweise war der klassische KEAP1-Partner NRF2, oft als Haupttreiber antioxidativer Abwehr betrachtet, nicht für das Abtöten verantwortlich; tatsächlich machte das Entfernen von NRF2 die Zellen noch empfindlicher, was unterstreicht, dass MCB-613 einen anderen, nicht-kanonischen Weg ausnutzt.
Was das für künftige Behandlungen bedeuten könnte
MCB-613 selbst ist derzeit ein Werkzeugmolekül mit chemischen Nachteilen, die es ungeeignet als Medikament machen. Es zeigt jedoch ein kraftvolles Konzept: Wenn Lungenkrebse Resistenzen gegen EGFR-Inhibitoren entwickeln, können sie in einen belasteten Zustand eingespannt werden, der selektiv durch Verbindungen angegriffen werden kann, die KEAP1 in dysfunktionale Assemblies zwingen. Prinzipiell könnten verfeinerte Versionen solcher "molekularen Brücken" entwickelt werden, die sicherer und präziser sind und Onkologen ein Mittel geben, Tumoren in eine „unmögliche Wahl“ zu treiben zwischen Empfindlichkeit gegenüber der ursprünglichen Zieltherapie und Empfindlichkeit gegenüber einem nachfolgenden stressinduzierenden Wirkstoff. Diese evolutionsbasierte Einkreisungsstrategie könnte schließlich helfen, Resistenzen beim EGFR-mutierten Lungenkrebs — und möglicherweise auch bei anderen schwer behandelbaren Tumoren — zu verzögern oder zu überwinden.
Zitation: Bassil, C.F., Dillon, K., Anderson, G.R. et al. EGFR inhibitor-resistant lung cancers exhibit collateral sensitivity to a covalent, cysteine-independent KEAP1 oligomerizing molecular bridge.
Nat Commun17, 1726 (2026). https://doi.org/10.1038/s41467-026-68424-1