Clear Sky Science · de
Simultanes epigenomisches Profiling und Messung regulatorischer Aktivität mit e2MPRA
Die versteckten Schalter der Zelle lesen
Jede Zelle in Ihrem Körper trägt im Wesentlichen dieselbe DNA, dennoch verhalten sich eine Gehirnzelle und eine Leberzelle sehr unterschiedlich. Das Geheimnis liegt in kurzen DNA-Abschnitten, die wie Dimmer für Gene wirken und deren Aktivität hoch-, runter- oder ausschalten. Diese Studie stellt ein kraftvolles neues Werkzeug vor, genannt e2MPRA, mit dem Wissenschaftler tausende dieser Schalter gleichzeitig testen können, während sie zugleich beobachten, wie die DNA in der Zelle verpackt und markiert ist — entscheidende Schritte, um Entwicklung, Krankheitsrisiken und die Frage zu verstehen, warum manche genetische Varianten relevant sind und andere nicht.
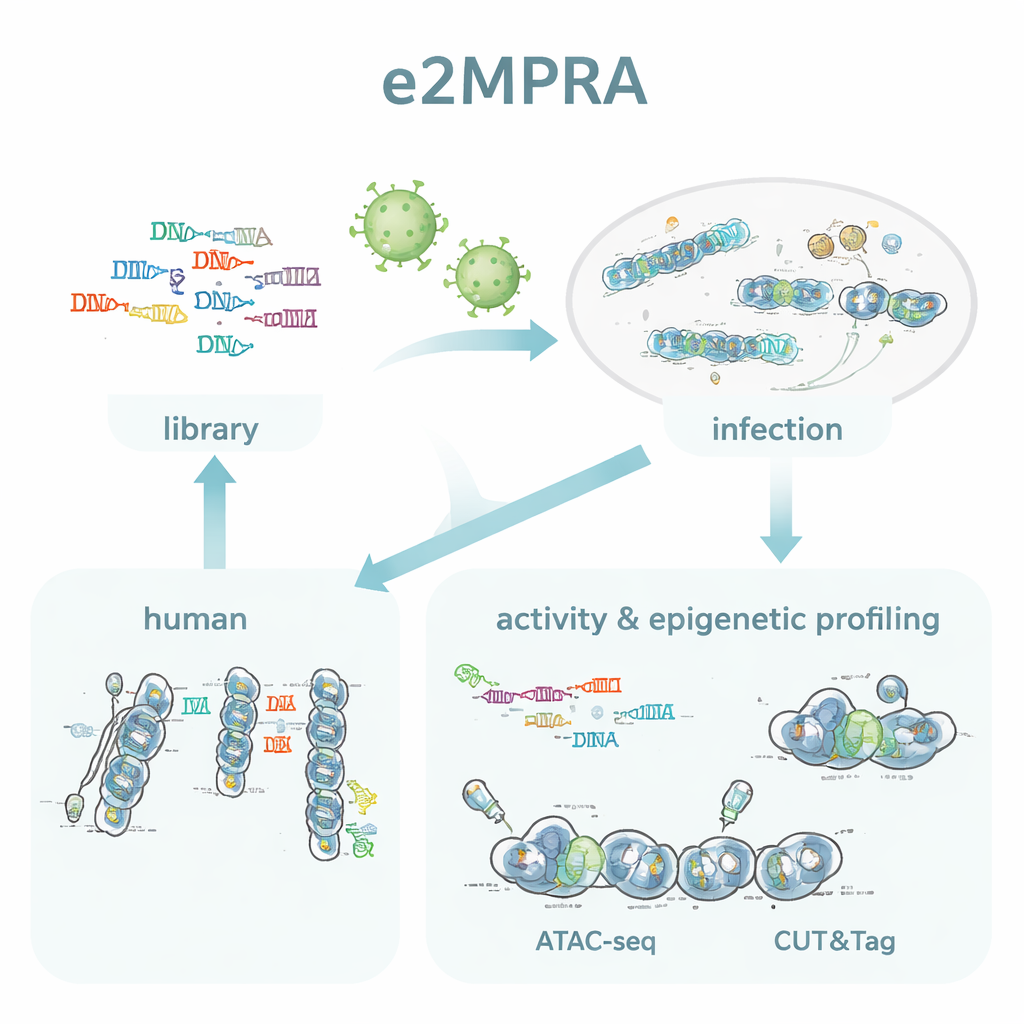
Von DNA-Karten zur DNA-Funktion
In den letzten zehn Jahren haben Forscher umfangreiche Karten „kandidater“ regulatorischer Schalter erstellt, bekannt als cis-regulatorische Elemente. Sie nutzen Methoden, die zeigen, wo DNA locker verpackt ist (offene Chromatinbereiche), welche Proteine gebunden sind und welche Histonmarken die umliegende DNA schmücken. Diese Karten sind beeindruckend, bleiben aber größtenteils beschreibend: nur weil ein Protein bindet oder eine Markierung erscheint, beweist das nicht, dass eine Sequenz tatsächlich die Genaktivität steuert. Traditionelle massiv-parallele Reporter-Assays (MPRAs) können testen, ob tausende DNA-Schnipsel einen Reportergen verstärken oder stilllegen, liefern jedoch keine Informationen darüber, welche Proteine dort binden oder welche epigenetischen Veränderungen mit dieser Aktivität einhergehen.
Ein Zweifach-Assay für Aktivität und epigenetische Marken
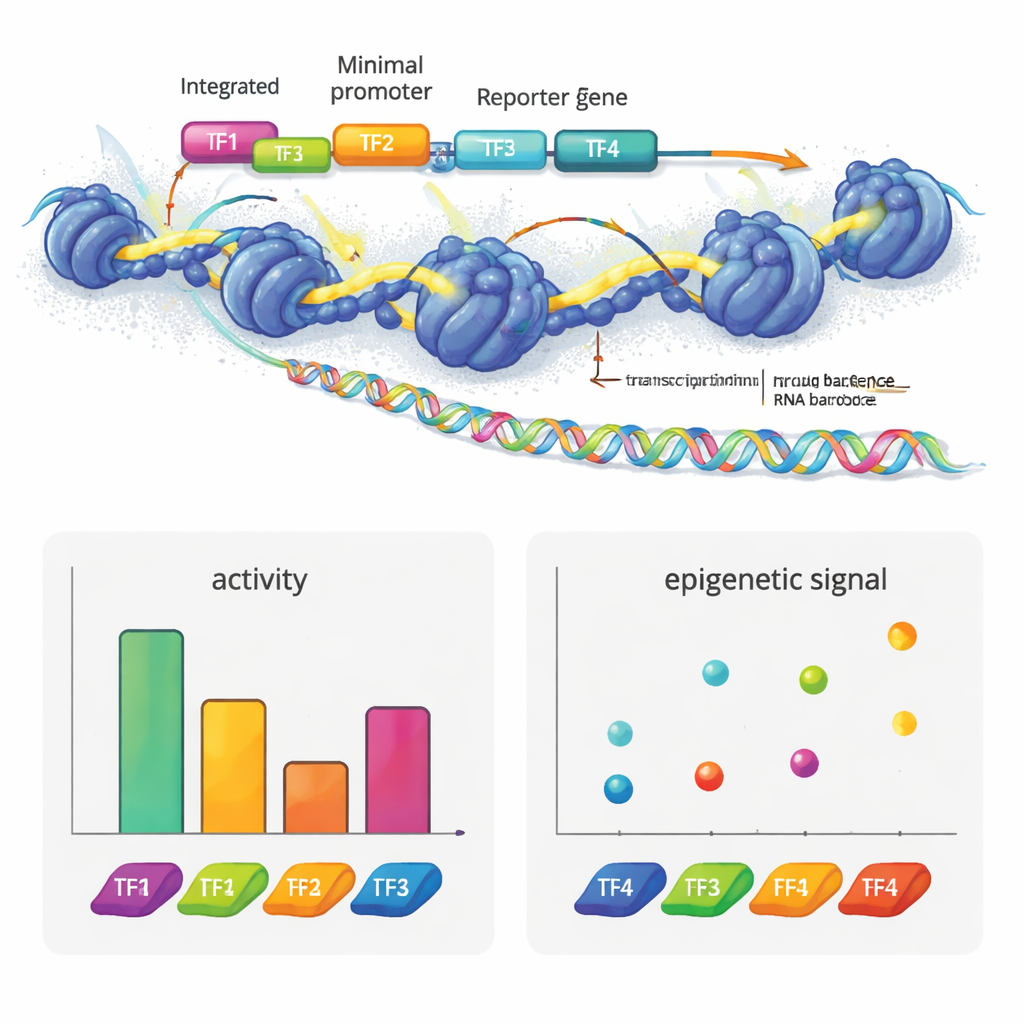
Die Autorinnen und Autoren entwickelten e2MPRA, indem sie ein lentivirales MPRA mit zwei epigenomischen Techniken, ATAC-seq und CUT&Tag, kombinierten. Zuerst bauen sie eine DNA-Bibliothek, in der jede Kandidaten-Regulatorsequenz mit einem kurzen Barcode verknüpft und neben einen minimalen Promotor und ein Reportergen gesetzt wird. Diese Bibliothek wird dann in Lentiviren verpackt und vielfach in die Genome kultivierter menschlicher Zellen integriert. Aus demselben Zellpool lesen sie Barcodes in RNA, um regulatorische Aktivität zu messen, und verwenden ATAC-seq sowie CUT&Tag, um die Chromatinöffnungsrate und eine wichtige Histonmarke (H3K27ac) gezielt an den eingefügten Sequenzen zu messen. Durch Normalisierung dieser Signale auf die tatsächliche Integrationshäufigkeit jeder Sequenz erhalten sie nebeneinander stehende Messungen von "Ein/Aus-Stärke" und epigenetischem Zustand für tausende Elemente in einem einzigen Experiment.
Entschlüsselung, wie Transkriptionsfaktor-Motive zusammenwirken
Um zu zeigen, was diese Technologie enthüllen kann, baute das Team synthetische Enhancer aus bekannten Transkriptionsfaktor-Motiven, die in Leberzellen verwendet werden. Sie ordneten diese Motive in unterschiedlicher Anzahl und Reihenfolge auf neutralen DNA-Vorlagen an. Einige Faktoren, wie HNF1A und XBP1, agierten als klassische Aktivatoren: Mehr Kopien erhöhten die Reporterexpression. Andere, wie HNF1A und ONECUT1, steigerten hauptsächlich die Chromatinzugänglichkeit und passten damit zu ihrer Rolle als "Pioneer"-Faktoren, die verschlossene DNA öffnen. PPARA zeigte ein anderes Muster: es veränderte stark Chromatin und Histonmarken, löste aber allein keine Transkription aus, kooperierte jedoch mit anderen Faktoren, um die Genaktivität zu steigern. REST, ein bekannter Repressor, dämpfte die Aktivität, wenn es neben Aktivatoren platziert wurde. Auffällig war, dass bereits das einfache Umordnen von vier Motiven die Enhancer-Stärke erheblich verändern konnte, und Aktivatoren taten sich tendenziell am besten, wenn sie näher am Promotor lagen — ein Hinweis auf eine Art grammatikalischer Struktur in der Anordnung dieser Motive.
Feinbestimmung sensibler Basen in Pluripotenz-Enhancern
Die Forschenden richteten sich anschließend auf Enhancer, die für die Identität von Stammzellen wichtig sind, und konzentrierten sich auf Regionen, die von den Pluripotenzfaktoren POU5F1 (auch OCT4 genannt) und SOX2 gebunden werden. Sie erzeugten dichte Mutationsbibliotheken, in denen jede Base von 100-Basen-Paar-Enhancern systematisch verändert wurde, sowie kleine sechsbisbasige Fenster, die zufällig vertauscht wurden. Mithilfe von e2MPRA in induzierten pluripotenten Stammzellen konnten sie erkennen, welche Mutationen die Reporteraktivität abschwächten oder verstärkten und wie sich dies auf Chromatinöffnung und Acetylierung auswirkte. Die Störung des POU5F1::SOX2-Bindungsmotivs reduzierte häufig sowohl Genaktivität als auch epigenetische Marken und bestätigte damit seine zentrale Rolle. In einem gut untersuchten Enhancer in der Nähe des POU5F1-Gens fanden sie außerdem, dass die Veränderung eines YY1-Motivs die Transkription erhöhte, aber die Chromatinöffnung verringerte — ein Hinweis darauf, dass manche Faktoren einen offenen Zustand fördern können und zugleich die Expression einschränken.
Was das für Gene und Krankheit bedeutet
e2MPRA bildet nicht alle Nuancen der natürlichen 3D-Vernetzung des Genoms perfekt nach, bietet jedoch eine praktikable Möglichkeit, tausende regulatorische Sequenzen und Varianten unter gleichen Bedingungen zu vergleichen. Indem es eine funktionelle Messung (wie stark ein DNA-Segment ein Gen antreibt) mit epigenetischen Messungen (wie diese DNA verpackt und markiert ist) koppelt, hilft diese Methode zu erklären, warum bestimmte Transkriptionsfaktor-Motive, Kombinationen und Positionen so entscheidend sind. Langfristig können Werkzeuge wie e2MPRA die Interpretation nichtkodierender genetischer Varianten, die mit Krankheiten verbunden sind, erleichtern, das Design synthetischer Enhancer für Gentherapie verbessern und zum Aufbau eines umfassenderen "Regulationscodes" beitragen, der DNA-Sequenz mit Zellverhalten verbindet.
Zitation: Zhang, Z., Georgakopoulos-Soares, I., Bourque, G. et al. Simultaneous epigenomic profiling and regulatory activity measurement using e2MPRA. Nat Commun 17, 1724 (2026). https://doi.org/10.1038/s41467-026-68422-3
Schlüsselwörter: Genregulation, Enhancer, Epigenetik, Transkriptionsfaktoren, funktionelle Genomik