Clear Sky Science · de
Umfassende Kartierung der Dynamik und Wechselwirkungen von RNA-Modifikationen mittels Deep Learning und Nanopore-Direktrna-Sequenzierung
Die verborgenen Satzzeichen der RNA
Die RNA-Moleküle unserer Zellen sind keine einfachen Ketten aus A, C, G und U. Sie sind mit Dutzenden winziger chemischer Markierungen versehen, die wie Satzzeichen wirken und steuern, welche Gene angeschaltet werden, wie Proteine hergestellt werden und wie Zellen auf Stress oder Krankheit reagieren. Bislang konnten Forschende diese Markierungen meist nur einzeln untersuchen, wodurch es schwer war, ihr Zusammenspiel im ganzen Genom zu erkennen. Dieser Artikel stellt ORCA vor, ein Deep‑Learning-System, das native RNA-Moleküle direkt liest und eine globale, mehrschichtige Karte dieser chemischen Markierungen und ihrer Wechselwirkungen erstellt.
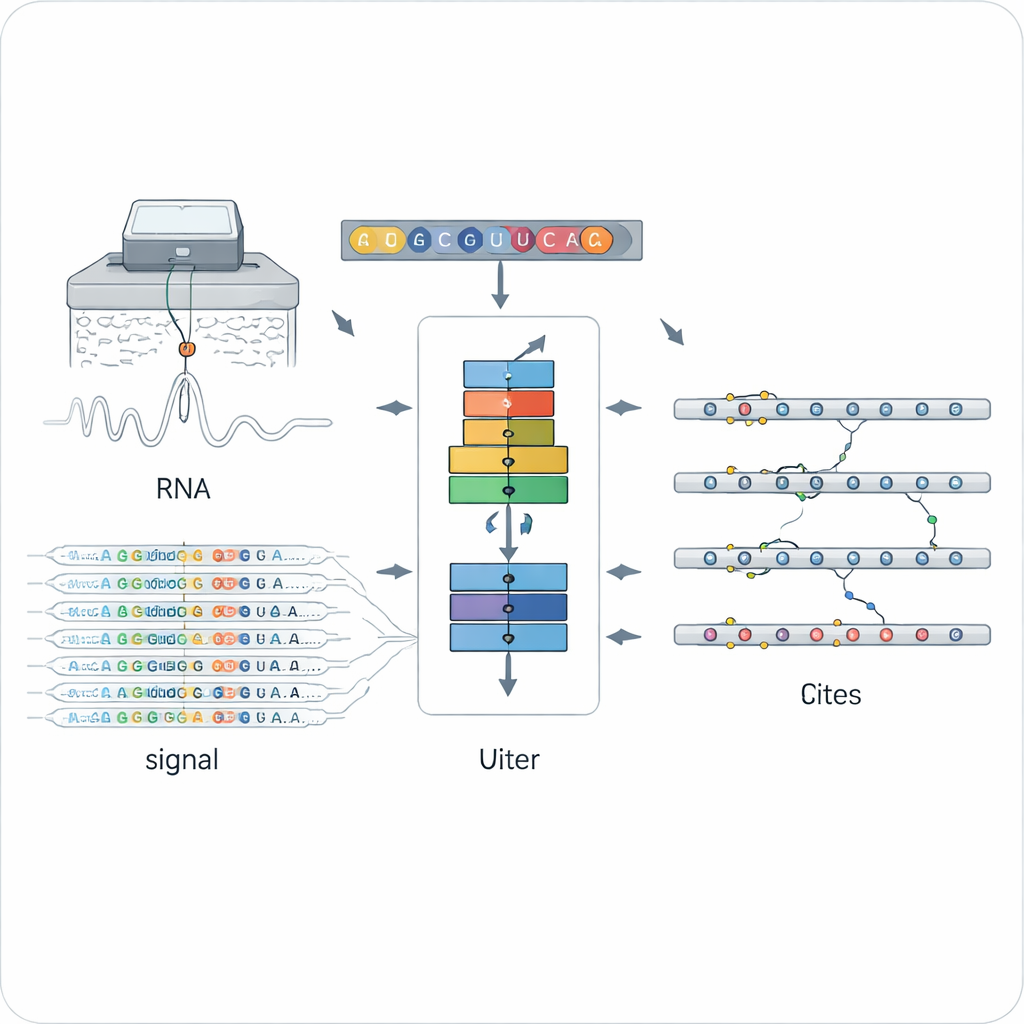
Eine neue Methode, chemische Markierungen auf RNA zu lesen
Traditionelle Methoden zur Erkennung von RNA-Modifikationen beruhen meist auf speziellen Antikörpern oder auf chemischen Verfahren, die auf einen einzigen Markierungstyp zugeschnitten sind, wie etwa das weitverbreitete N6‑Methyladenosin (m6A). Das macht sie leistungsfähig, aber eng: Jede Methode detektiert nur eine Art von Markierung und oft nur in einem bestimmten experimentellen Setup. Die Nanopore-Direktrna-Sequenzierung öffnete einen anderen Weg, indem einzelne RNA-Moleküle durch eine winzige Pore gezogen und Änderungen im elektrischen Strom gemessen werden, die von der genauen chemischen Struktur jeder Base abhängen. Modifizierte und unmodifizierte Buchstaben verzerren das Signal und das Basencalling auf subtil unterschiedliche Weise, aber aus diesen verrauschten, hochdimensionalen Daten über viele Modifikationstypen hinweg sinnvolle Informationen zu ziehen, war eine große Herausforderung.
Einem neuronalen Netz das Erkennen beliebiger Markierungen beibringen
ORCA (Omni‑RNA modification Characterization and Annotation) begegnet dieser Herausforderung in zwei Stufen. Zuerst fokussiert es sich auf ein kleines Fenster rund um jede Position in der RNA und aggregiert sowohl das rohe elektrische Signal als auch das Muster der Sequenzierfehler über viele Reads. Da nur ein Bruchteil der RNA-Kopien eine bestimmte Markierung trägt, zeigen echt modifizierte Stellen stärker verzerrte Signalausprägungen und häufiger auftretende Basecalling-Fehler an dieser Position. ORCA nutzt ein tiefes rekurrentes neuronales Netz, das mit einer „adversarialen“ Strategie trainiert wird, sodass es allgemeine Muster lernt, die modifizierte von unmodifizierten Stellen unterscheiden, ohne sich auf einen einzigen bekannten chemischen Typ festzulegen. So kann ORCA jeder Stelle einen Modifikationsscore und eine geschätzte Fraktion modifizierter Moleküle zuweisen.
Die Identität jeder Markierung erlernen
In der zweiten Stufe lernt ORCA, welche Art von chemischer Markierung vorliegt. Die Autoren füttern das Modell mit einer Reihe von hochzuverlässigen Stellen aus öffentlichen Datenbanken, bei denen konventionelle Experimente bereits m6A, 5‑Methylcytosin (m5C), Pseudouridin (Ψ), Inosin, 2′‑O‑Methylierung und mehrere seltenere Markierungen identifiziert haben. ORCA verdichtet die Signalprofile, den Sequenzkontext und kurze Sequenz„motive“ um jede Stelle in eine niedrigerdimensionale Karte und verfeinert sich dann, um den Modifikationstyp und die genaue Base vorherzusagen. Entscheidenderweise werden auch unbeschriftete Stellen als „Hintergrund“-Beispiele genutzt, was dem Modell hilft, unbekannte Markierungen nicht fälschlich einer bekannten Kategorie zuzuordnen. Nach dem Training kann ORCA diese gelernten Labels auf Zehntausende zuvor nicht annotierter Stellen im Transkriptom übertragen.
Viele Modifikationen gleichzeitig sehen
Bei Anwendung von ORCA auf menschliche und Maus-Zellen zeigen die Autoren, dass es nicht nur mit führenden Werkzeugen für spezifische Markierungen wie m6A, m5C und Ψ mithält oder sie übertrifft, sondern auch Markierungen erkennt, auf die es nie explizit trainiert wurde. Beispielsweise rekonstruierte ORCA die meisten unabhängig gemessenen m6A-Stellen korrekt, selbst wenn m6A-Daten während des Trainings zurückgehalten wurden, und unterschied sie von ähnlichen, unmodifizierten Sequenzmotiven. Ähnliches galt für 2′‑O‑Methylgruppen, Inosin‑Editing-Stellen und eine Vielzahl chemischer Veränderungen auf ribosomaler RNA, einschließlich vieler seltener Modifikationen, die mittels Massenspektrometrie nachgewiesen wurden. Insgesamt erweitert ORCA den bekannten Katalog an RNA-Modifikationsstellen erheblich, mit vielfachen Zuwächsen bei annotierten m5C-, Ψ-, m7G- und anderen niedrig abundant vorkommenden Markierungen im Vergleich zu bestehenden Datenbanken.
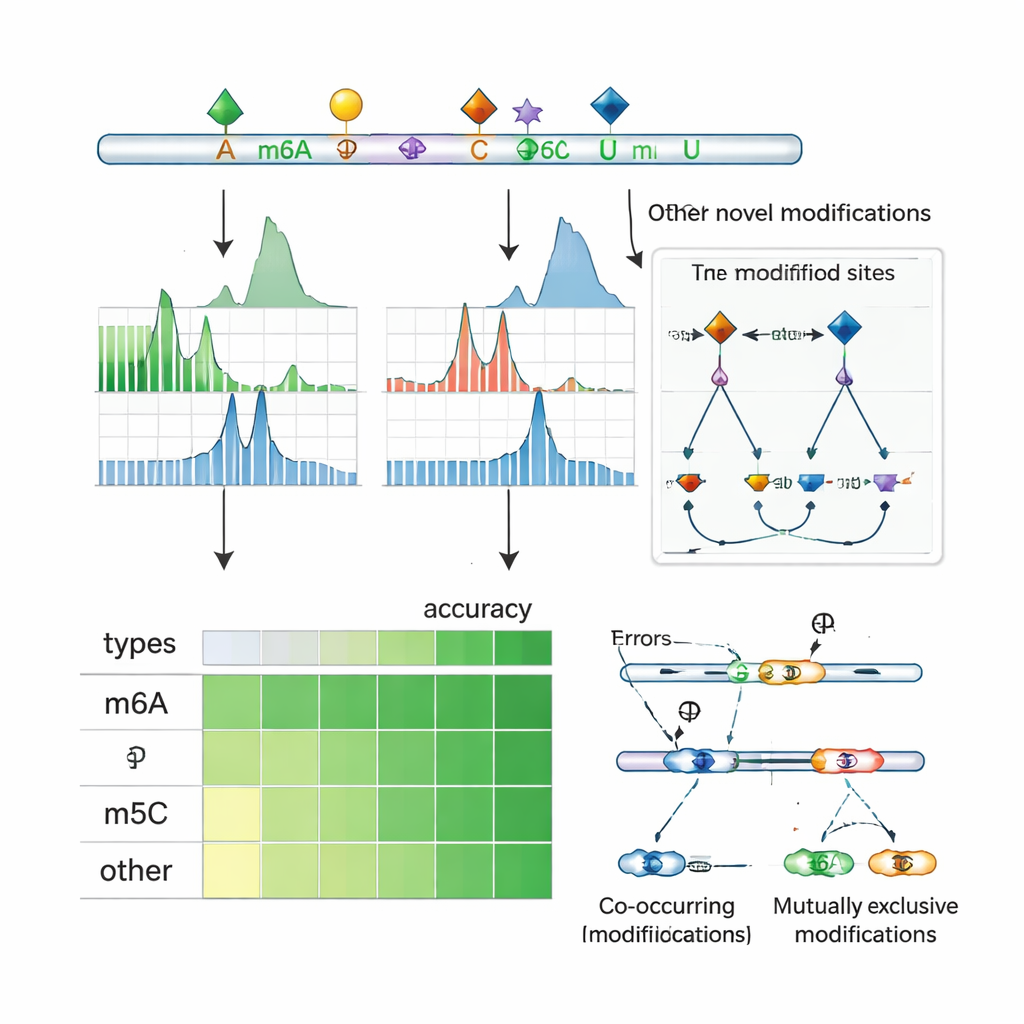
Wechselwirkungen und Kontrolle des Spleißens aufdecken
Da die Nanopore-Sequenzierung ganze RNA-Moleküle liest, kann ORCA untersuchen, welche Markierungen zusammen auf demselben Transkript vorkommen und welche sich gegenseitig ausschließen. Die Autoren clustern benachbarte Markierungen entlang von RNAs und verwenden ein probabilistisches Modell, um zu schlussfolgern, ob Paare von Stellen häufig gemeinsam modifiziert sind oder sich auf Einzelmolekülen gegenseitig ausschließen. Sie finden häufiges gemeinsames Auftreten von m6A mit m5C und anderen Markierungen sowie viele Regionen, in denen eine Stelle nur modifiziert ist, wenn die Nachbarstelle nicht modifiziert ist. In humanen Zelllinien liegen diese Muster oft in der Nähe von Exons, die alternativ ein- oder ausgeschlossen werden, und sie überlappen Bindungsstellen für Spleißregulatoren und „Reader“-Proteine, die modifizierte RNA erkennen. In spezifischen Genen zeigt ORCA, dass bestimmte Spleißvarianten für ein Muster von Markierungen angereichert sind, während alternative Varianten ein anderes Muster tragen, wodurch die lokale chemische Dekoration der RNA mit der Art und Weise verknüpft wird, wie Botschaften geschnitten und zusammengesetzt werden.
Warum das für Biologie und Medizin wichtig ist
Durch die Kombination von Direktrna-Sequenzierung mit flexiblem Deep Learning verwandelt ORCA ein kompliziertes elektrisches Signal in eine reichhaltige, mehrschichtige Karte chemischer Markierungen im Transkriptom. Für Nicht‑Spezialisten ist das wichtigste Ergebnis, dass Forschende nun nicht nur sehen können, wo einzelne RNA-Modifikationen auftreten, sondern wie viele verschiedene Markierungen dasselbe Molekül schmücken und wie diese Kombinationen mit Genregulation, insbesondere dem RNA‑Spleißen, zusammenhängen. Dieses Framework ermöglicht es, RNA‑„Epigenetik“ in vielen Zelltypen und Bedingungen zu untersuchen, ohne für jede Markierung ein neues Experiment entwerfen zu müssen, und ebnet den Weg für Entdeckungen darüber, wie diese winzigen chemischen Feinabstimmungen zur Entwicklung, Gehirnfunktion und zu Krankheiten wie Krebs und neurologischen Störungen beitragen.
Zitation: Dong, H., Gao, Y., Cai, Z. et al. Comprehensive mapping of RNA modification dynamics and crosstalk via deep learning and nanopore direct RNA-sequencing. Nat Commun 17, 1722 (2026). https://doi.org/10.1038/s41467-026-68419-y
Schlüsselwörter: RNA-Modifikationen, Nanopore-Sequenzierung, Deep Learning, Epitranskriptom, Alternatives Splicing