Clear Sky Science · de
Verwendung linearer Referenzen des Pangenoms zur Entdeckung fehlender Autismusvarianten
Warum versteckte DNA-Veränderungen bei Autismus wichtig sind
Die meisten Familien, die eine genetische Untersuchung für ein autistisches Kind anstreben, hoffen auf klare Antworten, doch bei etwa vier von fünf bleibt eine definitive genetische Erklärung aus. Diese Studie nimmt einen wichtigen Grund dafür in den Blick: Viele wirkungsvolle DNA-Veränderungen sind zu komplex, um von Standardtests erkannt zu werden. Indem die Forschenden nahezu vollständige Genome von 189 Personen aus 51 autismusbetroffenen Familien zusammenstellten und mit einer neuen, reichhaltigeren "Pangenom"-Referenz verglichen, zeigen sie, wie fortschrittliche Sequenzierung seltene, bislang unsichtbare Mutationen aufdecken kann, die zur Erklärung mancher Autismusfälle und verwandter Erkrankungen beitragen könnten.
Blick über Standardtests hinaus
Traditionelle klinische Tests basieren auf kurzen DNA-Fragmenten, um das Genom einer Person zu durchsuchen. Das funktioniert gut für viele einzelne Basenänderungen, versagt jedoch oft in repetitiven oder strukturell komplexen Regionen — genau dort, wo einige wirkungsvolle krankheitsverursachende Mutationen verborgen sind. Das Team konzentrierte sich auf Familien, bei denen frühere Kurzread-Genome-, Exom- oder Paneltests keine Ursache für Autismus- oder Rett-ähnliche Symptome gefunden hatten. Mit Langzeitsequenzierung, die deutlich längere DNA-Abschnitte liest, erstellten sie hochwertige, phasierte Genome für 189 Personen. Das ermöglichte, die beiden Chromosomenkopien jeder Person — eine von jeder Elternseite — mit sehr wenigen Lücken zu rekonstruieren.
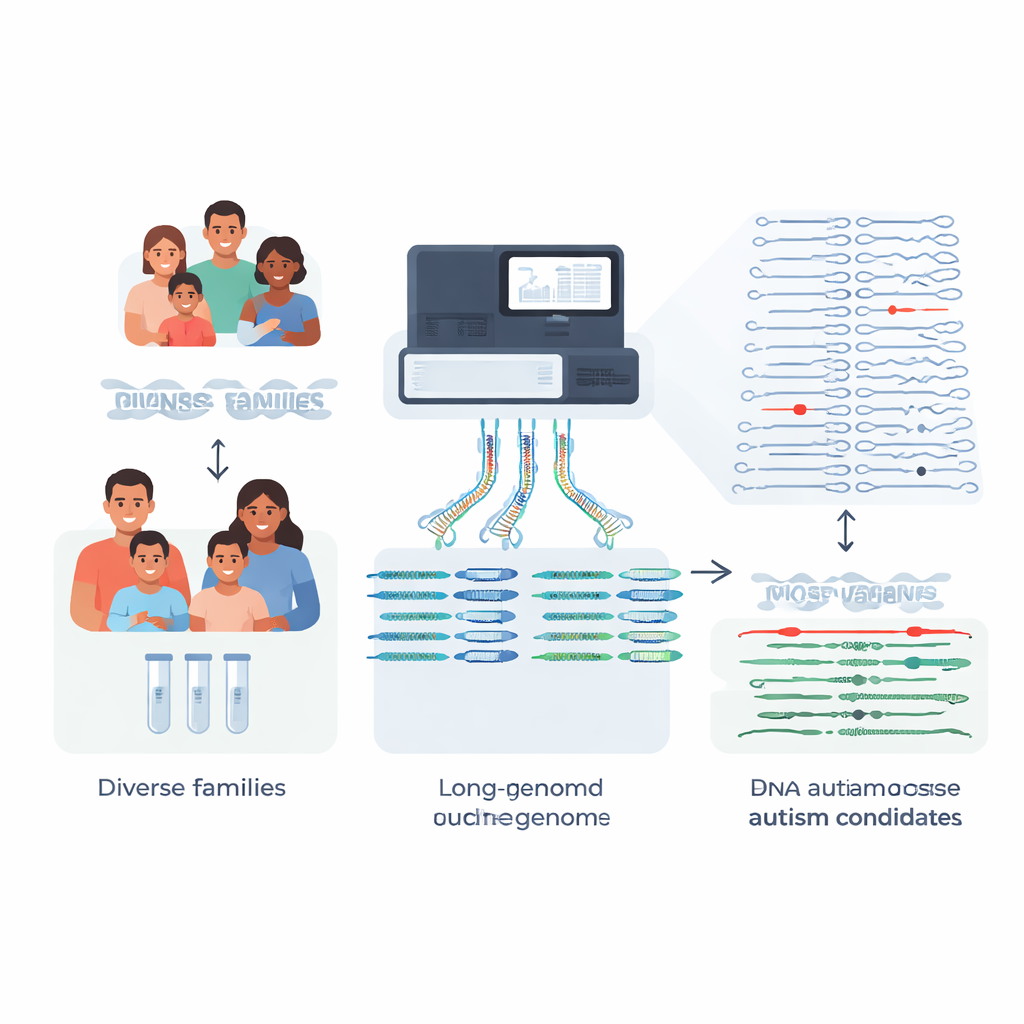
Strukturelle Varianten: große Veränderungen mit großer Wirkung
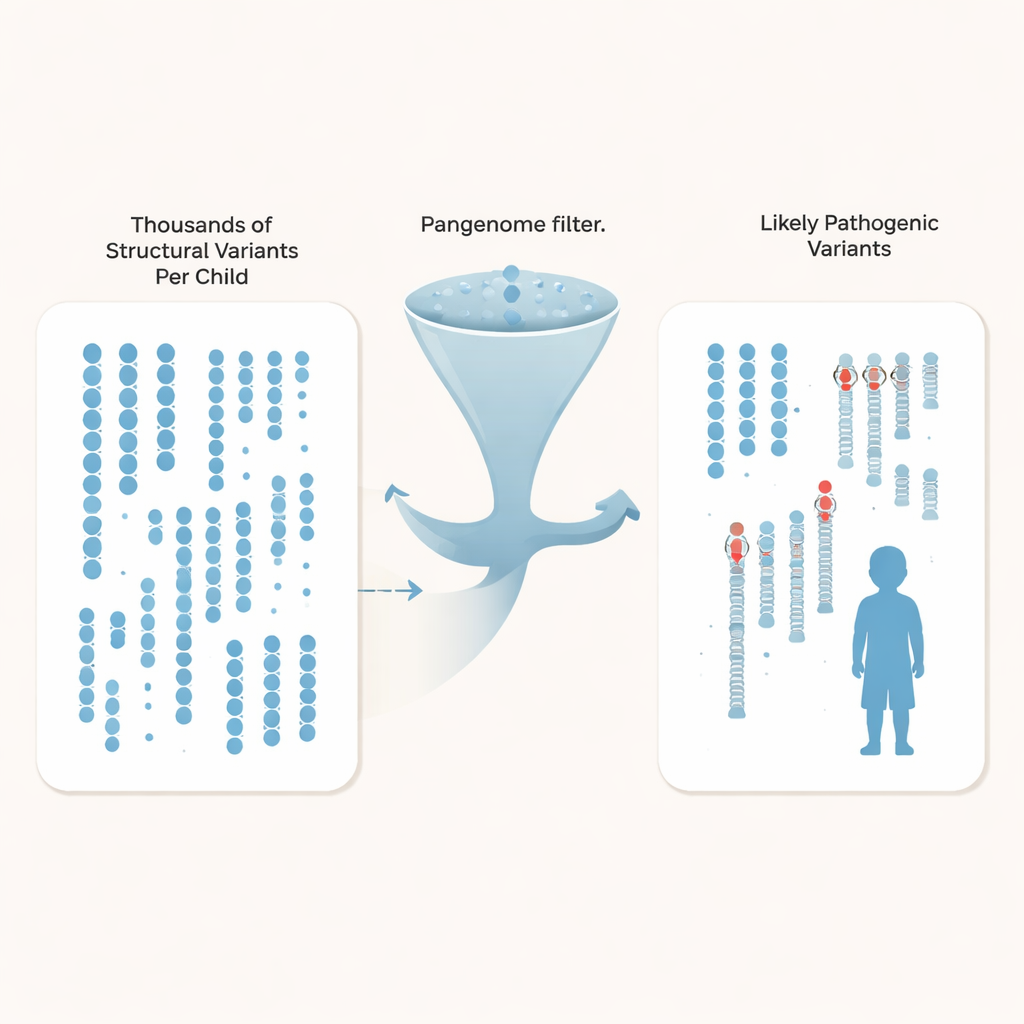
Statt nur einzelne Basenunterschiede zu verfolgen, richteten die Forschenden ihren Fokus auf strukturelle Varianten — Einfügungen, Löschungen und Umlagerungen, die mindestens 50 Basen betreffen und Gene oder deren Steuerungsbereiche stören können. Jedes Kind trug rund 27.000 solcher Varianten, doch die große Mehrheit sind harmlose, in der Bevölkerung verbreitete Unterschiede. Durch den Vergleich ihrer Autismusfamilien mit Hunderten tief sequenzierter Pangenom-Kontrollgenomen aus unterschiedlichen Abstammungen konnte das Team für jedes Kind über 97 % der häufigen strukturellen Varianten herausfiltern, sodass pro Genom etwa 600 seltene Kandidaten übrig blieben und bei der größten Kontrollgruppe sogar nur noch etwa 200.
Entdeckung übersehener Mutationen in bekannten Risikogenen
Mit dem drastisch verkleinerten Suchraum integrierten die Autorinnen und Autoren mehrere Evidenzlinien: bekannte Autismus- und neuroentwicklungsstörungs-Gene, regulatorische Regionen, die im sich entwickelnden menschlichen Kortex aktiv sind, und Vererbungsmuster innerhalb jeder Familie. Sie fanden drei eindeutig pathogenetische Mutationen, die frühere Tests übersehen hatten. Dazu gehörte ein neues Stoppsignal im SYNGAP1-Gen, das für die Synapsenfunktion wichtig ist, und eine Deletion, die das letzte Exon von MECP2 entfernt — einem zentralen Rett-Syndrom-Gen — obwohl die Patientin bereits mehrfach klinisch getestet worden war. Außerdem bestätigten sie eine krankheitsverursachende Veränderung in TBL1XR1, einem Gen, das mit MECP2 interagiert. Insgesamt hoben sie neun zusätzliche strukturelle Varianten hervor — häufig vererbt und in regulatorischen Regionen in der Nähe gehirnrelevanter Gene gelegen — als starke Kandidaten für zukünftige funktionelle Tests.
Was die Studie nicht fand — und warum das trotzdem wichtig ist
Trotz dieser tiefgehenden Suche beobachteten die Autorinnen und Autoren keinen klaren allgemeinen Überschuss an strukturellen Varianten bei autistischen Kindern im Vergleich zu ihren nicht betroffenen Geschwistern, zumindest nicht in dieser vergleichsweise kleinen Stichprobe. Es gab jedoch einen Hinweis auf mehr strukturelle Veränderungen auf dem X‑Chromosom bei betroffenen Mädchen, und die nahezu vollständigen X‑ und Y‑Assemblies ermöglichten es, ungewöhnliche Muster wie extreme Schieflagen der X‑Chromosomen-Inaktivierung zu erkennen. Solche Merkmale könnten wichtige Hinweise liefern, sobald mehr Familien untersucht werden. Entscheidend ist: Die Arbeit zeigt, dass Langzeitlektüre krankheitsverursachende Varianten wiederfinden kann, die Kurzread-Methoden übersehen — insbesondere in schwierigen Bereichen des Genoms und in Kontrollregionen, die die Genaktivität feinjustieren.
Was das für Familien und zukünftige Tests bedeutet
Für Familien ist die unmittelbare Auswirkung zwar bescheiden, aber bedeutsam: Bei diesen schwer zu klärenden Fällen erhielten etwa 6 % eine klare genetische Diagnose, und fast jede fünfte Familie bekam starke neue Kandidatenvarianten zur weiteren Untersuchung. Für das Fachgebiet ist die Botschaft größer. Wenn dem Pangenom mehr diverse, vollständige Referenzgenome hinzugefügt werden und Langzeitsequenzierung leichter zugänglich wird, werden Kliniker gängige strukturelle Varianten schneller ausschließen und sich rasch auf eine kleine Anzahl seltener, potenziell schädlicher Varianten pro Patient konzentrieren können. Dieser Wandel könnte nach und nach viele der heutigen ungelösten Autismusfälle in solche verwandeln, bei denen die zugrunde liegende Biologie — und mögliche Wege zu Unterstützung und Behandlung — deutlich besser verstanden sind.
Zitation: Sui, Y., Lin, J., Noyes, M.D. et al. Using the linear references from the pangenome to discover missing autism variants. Nat Commun 17, 1681 (2026). https://doi.org/10.1038/s41467-026-68378-4
Schlüsselwörter: Genetik des Autismus, Langzeit-Lesung, Strukturelle Varianten, menschliches Pangenom, Rett-Syndrom