Clear Sky Science · de
Belege über Arten hinweg für eine entwicklungsbedingte Ursache von erwachsener Hypersomnie durch Verlust synaptischer Adhäsionsmoleküle beat-Ia/CADM2
Warum zu viel Schlaf ein ernstes Problem sein kann
Viele Menschen beneiden schwere Schläfer, doch für Betroffene mit idiopathischer Hypersomnie kann überwältigende Schläfrigkeit Arbeit, Schule und Beziehungen zerstören. Diese Erkrankung lässt Menschen nach einer vollen Nacht Ruhe erschöpft zurück, und Ärztinnen und Ärzte verstehen noch nicht vollständig, warum. Die vorliegende Studie kombiniert humangenetische Befunde mit Experimenten an Fruchtfliegen und Zebrafischen, um übermäßige Schläfrigkeit auf die Art und Weise zurückzuverfolgen, wie Hirnschaltkreise während der Entwicklung verdrahtet werden – und weist so auf eine mögliche neue Behandlungsstrategie hin.
Schläfrigkeitsgene in menschlicher DNA finden
Die Forschenden begannen damit, riesige genetische Datensätze von Hunderttausenden Menschen zu untersuchen, die von übermäßiger Schläfrigkeit tagsüber oder häufigem Nickerchen berichteten. Anstatt automatisch das dem Risiko-Variant nächstgelegene Gen als relevant anzunehmen, betrachteten sie größere 3D-Nachbarschaften des Genoms, sogenannte „topologische Domänen“, um alle plausiblen Gene zu erfassen. Anschließend nutzten sie Rechenwerkzeuge, um passende Gene in Fruchtfliegen zu finden und landeten bei mehr als 200 Fliegengenen, die getestet wurden. Indem sie diese Gene systematisch in allen Neuronen herunterregulierten, suchten sie nach Fliegen, die deutlich mehr schliefen als normal. Zu den stärksten Treffern gehörten Gene aus der „beaten path“-Familie, Gegenstücke des menschlichen Gens CADM2, das ein Molekül codiert, das Nervenzellen hilft, an Synapsen zusammenzuhalten und Verbindungen zu bilden. 
Schläfrige Fliegen und schläfrige Fische
Wurde die fliegenspezifische Version von CADM2, genannt beat-Ia, in Neuronen reduziert, schliefen die Fliegen deutlich länger bei Tag und Nacht. Sie wirkten beim Wachsein nicht träge; stattdessen waren ihre Schlafphasen länger, sie waren schwerer zu wecken und fielen nach Lichtanbruch schneller wieder in den Schlaf – Merkmale, die der menschlichen Hypersomnie nahekommen. Das Team testete CADM2 weiter in Zebrafischen, einem kleinen Wirbeltier, dessen Schlaf sich per Video verfolgen lässt. Die Störung des Fischgens cadm2b erhöhte den Schlaf der Fische, ohne ihre Bewegungen im Wachzustand zu verringern, was eine konservierte Rolle dieses Moleküls bei der Aufrechterhaltung von Wachheit stützt.
Wie frühe Verschaltungen das lebenslange Schlafverhalten formen
Eine zentrale Erkenntnis war, dass beat-Ia vor allem während der Gehirnentwicklung wichtig ist, nicht im Erwachsenenalter. Indem sie die Genherunterregulierung nur vor oder nur nach dem Schlüpfen der Fliegen aus dem Puppenstadium einschalteten, zeigten die Forschenden, dass die Störung von beat-Ia früh im Leben ausreicht, um lebenslange übermäßige Schläfrigkeit zu verursachen, während eine Reduktion nur bei erwachsenen Tieren kaum Wirkung hatte. Sie führten die Wirkung von beat-Ia auf eine kleine Gruppe von Neuronen zurück, die Neuropeptid F (NPF) produzieren, das Fliegenpendant zum vertebraten Neuropeptid Y (NPY). Bei normalen Fliegen senden NPF-Neurone dichte synaptische Projektionen in eine Hirnregion namens Suboesophagealer Bereich, wo sie auf spezifische inhibitorische (GABA-produzierende) Neurone treffen, die helfen, Wachheit zu stabilisieren. Bei Fliegen ohne beat-Ia bildeten sich die großen Synapsencluster in dieser Region nicht aus, obwohl die Nervenfasern physisch den Bereich erreichten. Das deutet darauf hin, dass fehlerhafte Synapsenbildung – und nicht grobe Fehlverdrahtung – das Gleichgewicht zugunsten übermäßigen Schlafs verschieben kann.
Von fehlverdrahteten Schaltkreisen zu einem Medikamentenziel
Mithilfe einer detaillierten Verschaltungskarte des Fliegengehirns identifizierte das Team einige wenige nachgeschaltete Neurone im suboesophagealen Bereich, die Eingaben von NPF-Zellen erhalten und als GABAerg vorhergesagt wurden. Diese Zellen stummzuschalten erhöhte den Schlaf, während ihre Aktivierung Wachheit förderte – ein Befund, der zur Idee passt, dass NPF normalerweise durch Ansteuerung eines lokalen inhibitorischen Netzwerks Wachheit aufrechterhält. Die Forschenden fragten dann, ob sich verlorene CADM2-ähnliche Funktion durch Verstärkung der NPY-Signalübertragung kompensieren lässt. Bei cadm2b-defizienten Zebrafischlarven normalisierte das Bad in einem Wirkstoff, der einen Untertyp des NPY-Rezeptors aktiviert (analog zum Fliegen-NPF-Rezeptor), den übermäßigen Schlaf, ohne normale Fische stark zu beeinflussen. Dieses artenübergreifende Resultat legt nahe, dass wenn synaptische Adhäsionsmoleküle während der Entwicklung aus dem Takt geraten, die Stärkung von NPY-Wegen im Erwachsenenalter helfen könnte, das richtige Schlaf-Wach-Gleichgewicht wiederherzustellen. 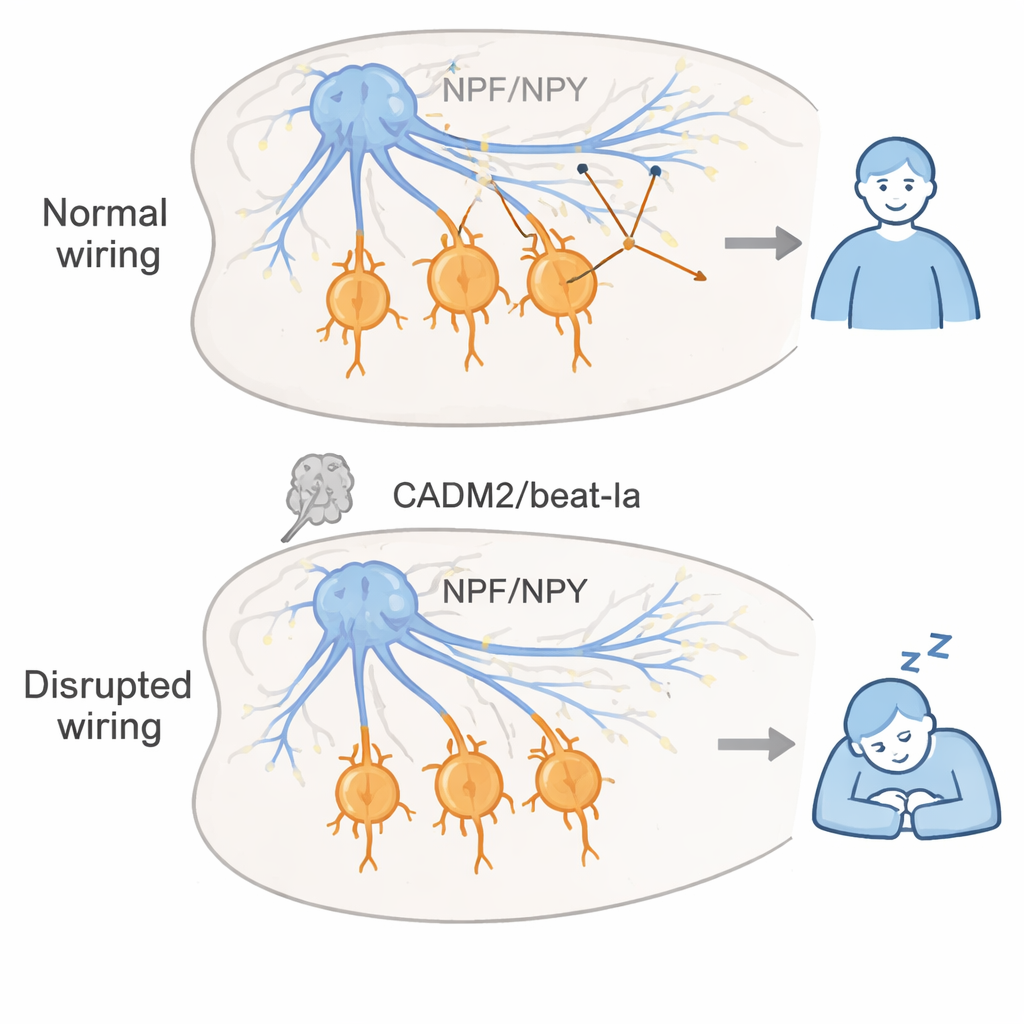
Was das für Menschen bedeutet, die nicht wach bleiben können
Insgesamt schlägt die Arbeit vor, dass einige Formen der idiopathischen Hypersomnie aus subtilen Fehlern in der Verdrahtung wachheitsfördernder Hirnschaltkreise in früher Entwicklungsphase stammen könnten, wobei CADM2 und verwandte Adhäsionsmoleküle beteiligt sind. Diese Veränderungen zerstören das Gehirn nicht, sondern rekonfigurieren die Stärke, mit der bestimmte Schlaf- und Erregungspfade miteinander kommunizieren. Wichtig ist, dass die Studie zeigt, dass selbst wenn das Verdrahtungsproblem in der Entwicklung beginnt, seine Folgen später noch behandelbar sein könnten, indem konservierte Neuropeptidsysteme wie NPY gezielt werden. Für Patientinnen und Patienten eröffnet das die Möglichkeit, dass zukünftige Medikamente, die diese Signalwege feinsteuern, präziser gegen die belastende Schläfrigkeit am Tag helfen könnten.
Zitation: Mace, K., Zimmerman, A., Chesi, A. et al. Cross-species evidence for a developmental origin of adult hypersomnia with loss of synaptic adhesion molecules beat-Ia/CADM2. Nat Commun 17, 1628 (2026). https://doi.org/10.1038/s41467-026-68343-1
Schlüsselwörter: idiopathische Hypersomnie, Schlafgenetik, synaptische Adhäsion, Neuropeptid Y, Gehirnentwicklung