Clear Sky Science · de
Molekulare Grundlage der Antagonisierung des dimeren menschlichen Arginin-Vasopressin-Rezeptors 1A
Warum ein hormoneller Gehirnrezeptor wichtig ist
Hormone wie Vasopressin und Oxytocin sind vor allem dafür bekannt, Wasserhaushalt, Blutdruck, Geburt und Bindungsverhalten zu steuern. Wie ihre Rezeptoren jedoch auf atomarer Ebene funktionieren, blieb weitgehend verborgen. Diese Studie zeigt detaillierte 3D-Strukturen eines Schlüsselrezeptors, des menschlichen Vasopressin-V1a-Rezeptors, der mit Sozialverhalten, Stress und mehreren Hirnerkrankungen in Verbindung steht. Das Verständnis seiner Form und der Mechanismen, mit denen Medikamente ihn blockieren, könnte Wissenschaftlern helfen, bessere Behandlungen für Erkrankungen wie Autismus, posttraumatische Belastungsstörung und Huntington-Krankheit zu entwickeln.
Ein Zwillingsrezeptor, der Herz-, Nieren- und Hirnsignale prägt
Der V1a-Rezeptor sitzt auf der Oberfläche vieler Zellen im Körper, besonders in Blutgefäßen, Nieren und bestimmten Hirnregionen. Wenn das Hormon Vasopressin an ihn bindet, aktiviert der Rezeptor intrazelluläre Signalwege, die Blutdruck, Flüssigkeitsbilanz und Hirnschaltkreise für soziale Interaktion, Emotion und Stress steuern. Genetische und klinische Studien haben abnormale V1a-Signale mit Autismus-Spektrum-Störungen, PTSD und Huntington-Krankheit verknüpft, was ihn zu einem attraktiven Wirkstoffziel macht. Mehrere V1a-blockierende Medikamente (Antagonisten) werden bereits verwendet oder in klinischen Studien geprüft, doch bis jetzt hatte noch niemand den menschlichen V1a-Rezeptor in hoher Auflösung gesehen, sodass wichtige Fragen zur Assemblierung und zur genauen Wirkweise dieser Medikamente offen blieben. 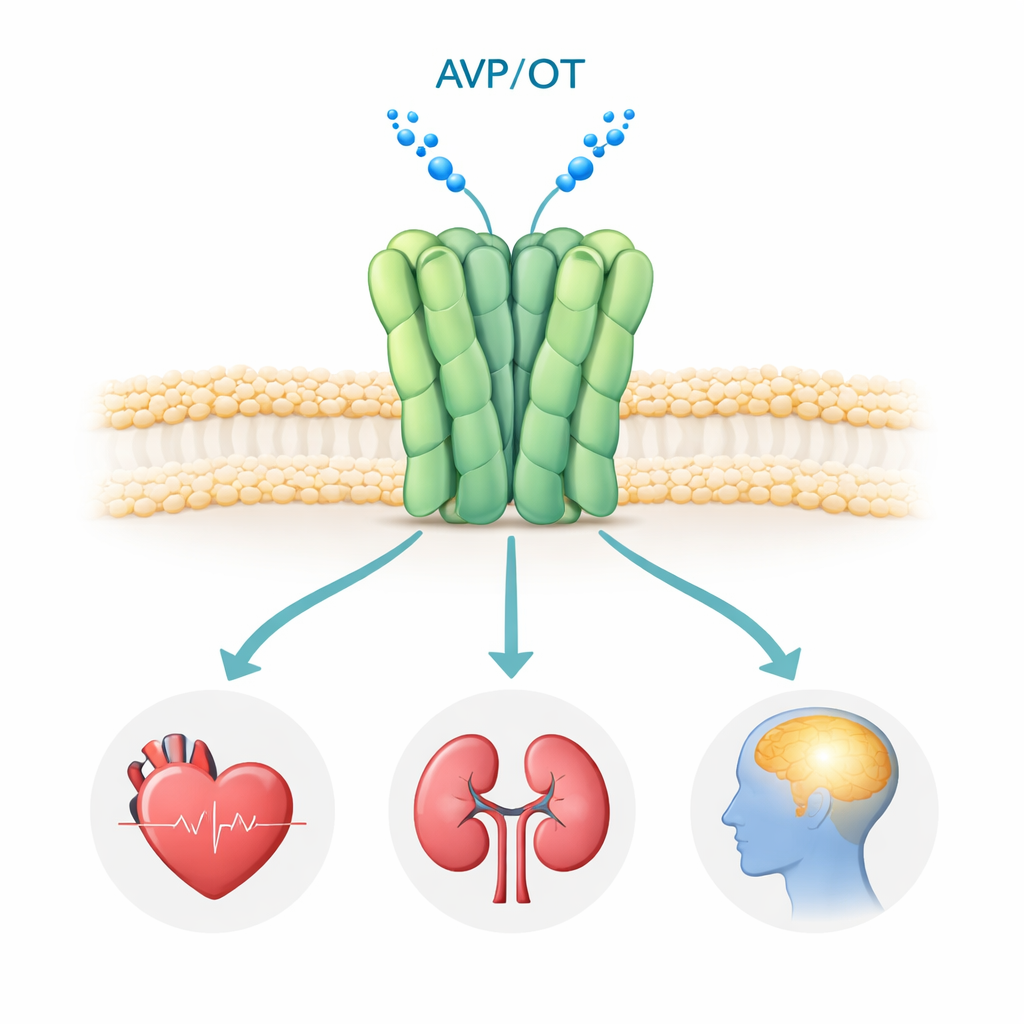
Den Rezeptor in mehreren medikamentengebundenen Zuständen einfrieren
Die Forschenden verwendeten Kryo-Elektronenmikroskopie, eine Technik, die Proteine blitzartig einfriert und mit einem Elektronenstrahl abbildet, um den menschlichen V1a-Rezeptor zu visualisieren. Um das Protein zu stabilisieren, konstruierten sie eine leicht modifizierte Form, die weiterhin Medikamente gut bindet, und kombinierten sie mit einem kleinen Antikörperfragment (Nanokörper) zur Bildgebung. Sie lösten Strukturen des Rezeptors allein und gebunden an drei medizinisch relevante Antagonisten: Atosiban (ein Peptidmedikament zur Verhinderung von Frühgeburten) sowie zwei gehirngängige Kleinmoleküle, Balovaptan und SRX246, die an Menschen mit Autismus oder Huntington-Krankheit getestet wurden. Alle Strukturen erreichten annähernd atomare Auflösung und zeigten die Positionen der sieben membranüberspannenden Helices des Rezeptors, flexibler Schleifen und der gebundenen Wirkstoffe.
Ein Rezeptor, der lieber paarweise arbeitet
Im Gegensatz zu verwandten Rezeptoren, die zuvor nur als Einzelmoleküle gesehen worden waren, erschien V1a in allen vier Kryo-EM-Strukturen als Paar—als Dimer. Die beiden Rezeptoren liegen nebeneinander in der Membran und stehen hauptsächlich über eine ihrer Helices in engem Kontakt, unterstützt durch polare und hydrophobe Wechselwirkungen. Um zu prüfen, ob diese Paarbildung auch in lebenden Zellen vorkommt, fusionierte das Team V1a mit einem hellen Fluoreszenzprotein und nutzte eine Einzelmolekül-Photobleaching-Methode: Wenn ein Fleck auf der Zelloberfläche zwei Rezeptorkopien enthielt, verschwand sein Licht in zwei Schritten. Etwa drei Viertel der beobachteten Flecken dunkelten genau in zwei Schritten ab, was die Vorstellung stützt, dass V1a auf natürliche Weise Dimere an der Zelloberfläche bildet. Wenn die Forschenden Schlüsselreste, die die Kontaktfläche bilden, mutierten, verschob sich der Rezeptor hin zu Einzelmolekülen und reagierte weniger auf Hormon und Medikament, was darauf hindeutet, dass das Dimer nicht nur eine strukturelle Erscheinung, sondern funktionell bedeutsam ist. 
Ein flexibles Tor am Hormonzugang
Das Team entdeckte eine unerwartete „Torklappe“ namens extrazelluläre Schleife 2 (ECL2), die oberhalb der Hormonbindetasche sitzt. Im medikamentenfreien (apo) Zustand liegt diese Schleife wie ein Deckel flach über der Tasche und bildet nicht die übliche Disulfidbrücke (eine Schwefel–Schwefel-Verbindung), wie sie bei vielen verwandten Rezeptoren zu sehen ist. Stattdessen falten sich Teile der Schleife in die Tasche und werden durch ein Netz von Wechselwirkungen mit umgebenden Helices gehalten, wodurch die große, klebrige Bindehöhle teilweise bedeckt wird. Wenn einer der drei Antagonisten bindet, schwingt ECL2 nach oben und weg, bildet die klassische Disulfidbrücke und schafft eine weite, mit Lösungsmittel gefüllte Höhlung, die die Medikamente einnimmt. Diese dramatische Bewegung legt nahe, dass V1a ECL2 als dynamische Barriere nutzen könnte, um zufällige Aktivierung durch Fremdmoleküle zu begrenzen, und dass Medikamente entweder die Schleife in ihrem flachen "Ruhezustand" einfangen oder ihre angehobene, offene Konformation ausnutzen könnten.
Wie drei Medikamente denselben Rezeptor auf unterschiedliche Weise stummschalten
Atosiban, das dem natürlichen Hormon Oxytocin sehr ähnlich ist, reicht von der Oberseite der Tasche bis zu ihrem Grund und verankert sich durch eine Kombination aus Wasserstoffbrücken und hydrophoben Kontakten. Durch die Veränderung weniger Schlüsselpositionen im Vergleich zu Oxytocin löst es jedoch die Verkettung interner Verschiebungen, die normalerweise für die Rezeptoraktivierung nötig sind, nicht aus: entscheidende „Mikroschalter“-Reste, die sich während der Signalübertragung bewegen, bleiben in ihren inaktiven Positionen verriegelt, die innere Höhlung, die das G-Protein aufnehmen würde, öffnet sich nicht, und eine für die Aktivierung wichtige Magnesiumbindestelle wird gestört. Im Gegensatz dazu sind Balovaptan und SRX246 kompakte, nicht-peptidische Moleküle, die tief in die Tasche eindringen, aber unterschiedliche Strategien nutzen. Balovaptan stützt sich auf einen starren, hydrophoben Kern, der sich dicht in eine tiefe Kluft einfügt, plus einen flexiblen polaren Schwanz, der zur Tascheneingang hinreicht. SRX246 verwendet eine modulare, fragmentartige Architektur, verankert durch einen β-Lactam-Kern, wobei verschiedene "Zonen" Subtaschen verschließen und sich zu den extrazellulären Schleifen erstrecken. In beiden Fällen stabilisieren die Medikamente eine inaktive Konformation, die mit der G-Protein-Bindung unvereinbar ist. Feine Unterschiede in Taschenform und Chemie—insbesondere an zwei Positionen auf den Helices 5 und 7—helfen zu erklären, warum Balovaptan und SRX246 V1a gegenüber eng verwandten Rezeptoren bevorzugen.
Folgen für künftige Therapien
Indem diese Arbeit hochauflösende Momentaufnahmen von V1a als Dimer liefert, eine zuvor ungesehene „flache“ Schleifenkonformation im medikamentenfreien Zustand aufzeigt und detailliert beschreibt, wie drei sehr unterschiedliche Antagonisten den Rezeptor ausschalten, gibt sie Wirkstoffentwicklern eine präzise strukturelle Landkarte zur gezielten Ansprache von V1a. Sie schlägt Wege vor, Wirkstoffe der nächsten Generation zu entwerfen, die entweder dimer-spezifische Merkmale ausnutzen oder den Rezeptor in einen besonders inaktiven Ruhezustand sperren—mit dem letztendlichen Ziel, Hirn- und Stress-assoziierte Erkrankungen gezielter und mit geringeren Nebenwirkungen zu behandeln.
Zitation: Zhong, P., Chu, B., Yu, Z. et al. Molecular basis of antagonism of the dimeric human arginine vasopressin receptor 1A. Nat Commun 17, 1622 (2026). https://doi.org/10.1038/s41467-026-68331-5
Schlüsselwörter: Vasopressin-V1a-Rezeptor, G-Protein-gekoppelter Rezeptor, Rezeptor-Dimerisierung, cryo-EM-Struktur, neuropsychiatrische Wirkstoffentwicklung