Clear Sky Science · de
Computergestützte Gestaltung allgemein einsetzbarer Cyclopropanasen mit stereodivergenter Selektivität
Warum winzige Dreiring‑Strukturen für Medikamente wichtig sind
Cyclopropane — dreiatomige Kohlenstoffringe — sind winzige, stark gespannte Bausteine, die das Verhalten eines Arzneistoffs im Körper dramatisch verändern können. Die genaue 3D‑Anordnung der Atome (ihre Stereochemie) entscheidet oft darüber, ob ein Molekül zu einem nützlichen Medikament wird oder ein inaktives beziehungsweise sogar schädliches Gegenstück bleibt. Dieser Artikel beschreibt eine computergestützte Strategie zur Konstruktion von Enzymen, die zuverlässig alle vier möglichen 3D‑Formen dieser Ringe aus denselben Ausgangsstoffen herstellen können und damit schnellere, sauberere Untersuchungen von Arzneimittelkandidaten ermöglichen.
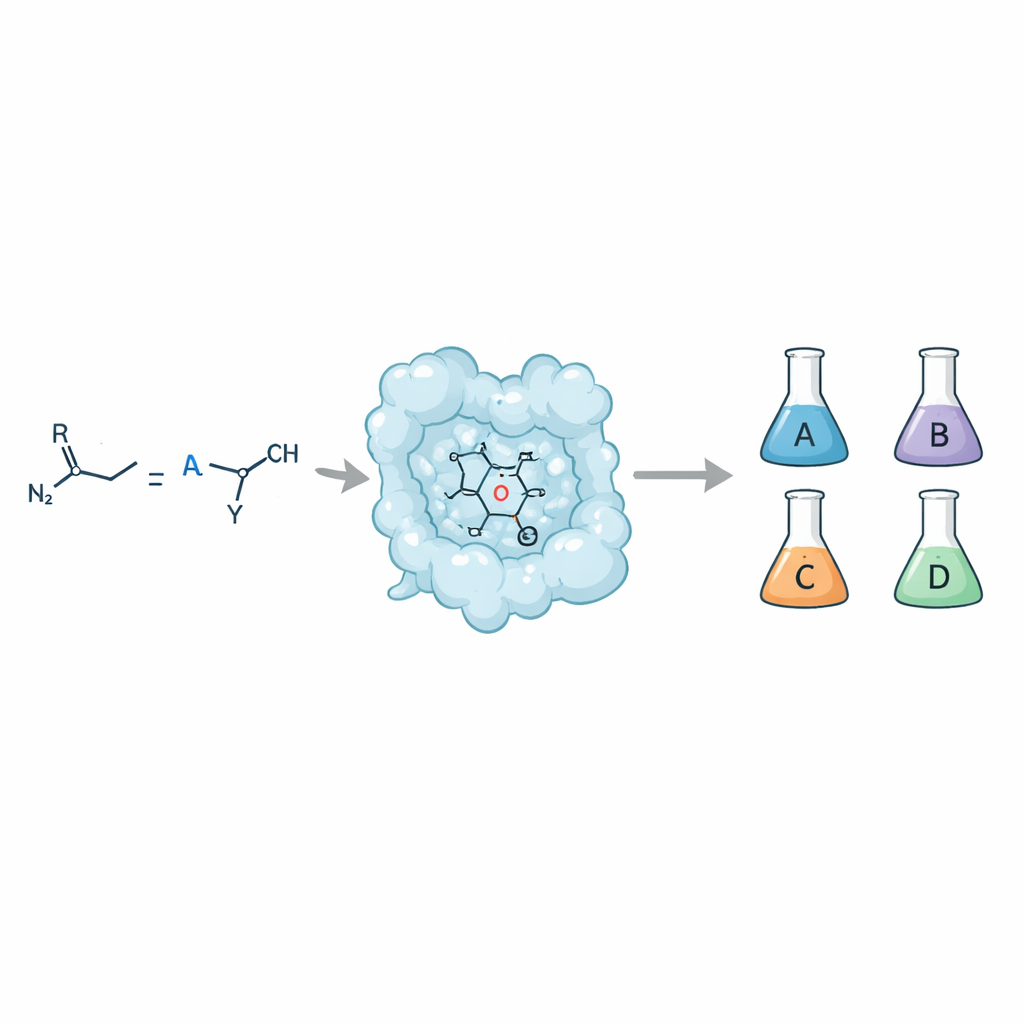
Von einem Rezept zu vier unterschiedlichen Ergebnissen
Wenn eine einfache Doppelbindung (ein Olefin) mit einem Carben‑Donor wie einer Diazoverbindung reagiert, kann sich ein Cyclopropanring bilden. Dieser Ring kann jedoch vier verschiedene stereoisomere Formen annehmen, die zwar dieselben Atome enthalten, aber unterschiedlich im Raum angeordnet sind. Chemiker möchten auf jede dieser Formen zugreifen, weil sie sehr unterschiedlich mit biologischen Zielmolekülen interagieren und wesentliche Eigenschaften von Arzneistoffen wie Resorption, Metabolismus und Sicherheit beeinflussen können. Traditionelle kleinmolekulare Katalysatoren erreichen diese Kontrolle manchmal, aber dies mit Enzymen — den Katalysatoren der Natur — zu realisieren war schwierig, insbesondere wenn sowohl hohe Selektivität als auch Breite gegenüber verschiedenen Substraten angestrebt werden.
Enzyme auf dem Computerbildschirm entwerfen
Die Autorinnen und Autoren entwickelten einen mechanistisch basierten, multistatischen computergestützten Arbeitsablauf, um dieses Problem zu lösen. Zuerst nutzten sie quantenchemische Berechnungen, um die flüchtigen Übergangszustände — die energiereichen Strukturen entlang des Reaktionspfads — für die Bildung jeder der vier Cyclopropan‑Stereoisomere zu modellieren. Diese Modelle wurden dann in die aktiven Zentren verschiedener hämhaltiger Proteine eingesetzt, und die Proteindesign‑Software Rosetta wurde verwendet, um zu bewerten, wie gut jedes Protein jeden Übergangszustand stabilisiert oder destabilisiert. Entscheidend war, dass der Design‑Score Mutationen belohnte, die sowohl den gewünschten Übergangszustand bevorzugten (positives Design) als auch die konkurrierenden Zustände benachteiligten (negatives Design) — praktisch eine Art Enzymschulung, damit es ein 3D‑Produkt den anderen vorzieht.
Ein vollständiges Enzym‑Werkzeugkasten aufbauen
Mit diesem Ansatz schuf das Team eine Familie von „Generalist“‑Cyclopropanasen. Ausgehend von Myoglobin gestalteten sie dessen aktives Zentrum so um, dass Varianten entstanden, die das trans‑(1R,2R)‑Cyclopropan mit sehr hoher Selektivität und guter Aktivität über mehr als 20 verschiedene Olefine liefern, einschließlich anspruchsvoller unaktivierter und elektronenschwacher Substrate. Ein zuvor entwickeltes Myoglobin lieferte bereits das komplementäre trans‑(1S,2S)‑Produkt. Um die beiden cis‑Produkte zu erhalten, wandten sich die Autorinnen und Autoren anderen Häm‑Proteinen zu. Sie remodelierten das bakterielle Enzym P450cam, um Varianten zu erhalten, die selektiv das cis‑(1S,2R)‑Produkt liefern, und sie nutzten das menschliche Indolamin‑2,3‑Dioxygenase‑1 (IDO1) — bislang nicht für Carben‑Chemie verwendet — um das cis‑(1R,2S)‑Produkt zu bevorzugen. Zusammengenommen können diese vier Biokatalysatoren jedes Stereoisomer desselben Cyclopropan‑Produktsets liefern, oft mit bis zu 99% Kontrolle sowohl über Diastereomer‑ als auch Enantiomerverhältnisse.
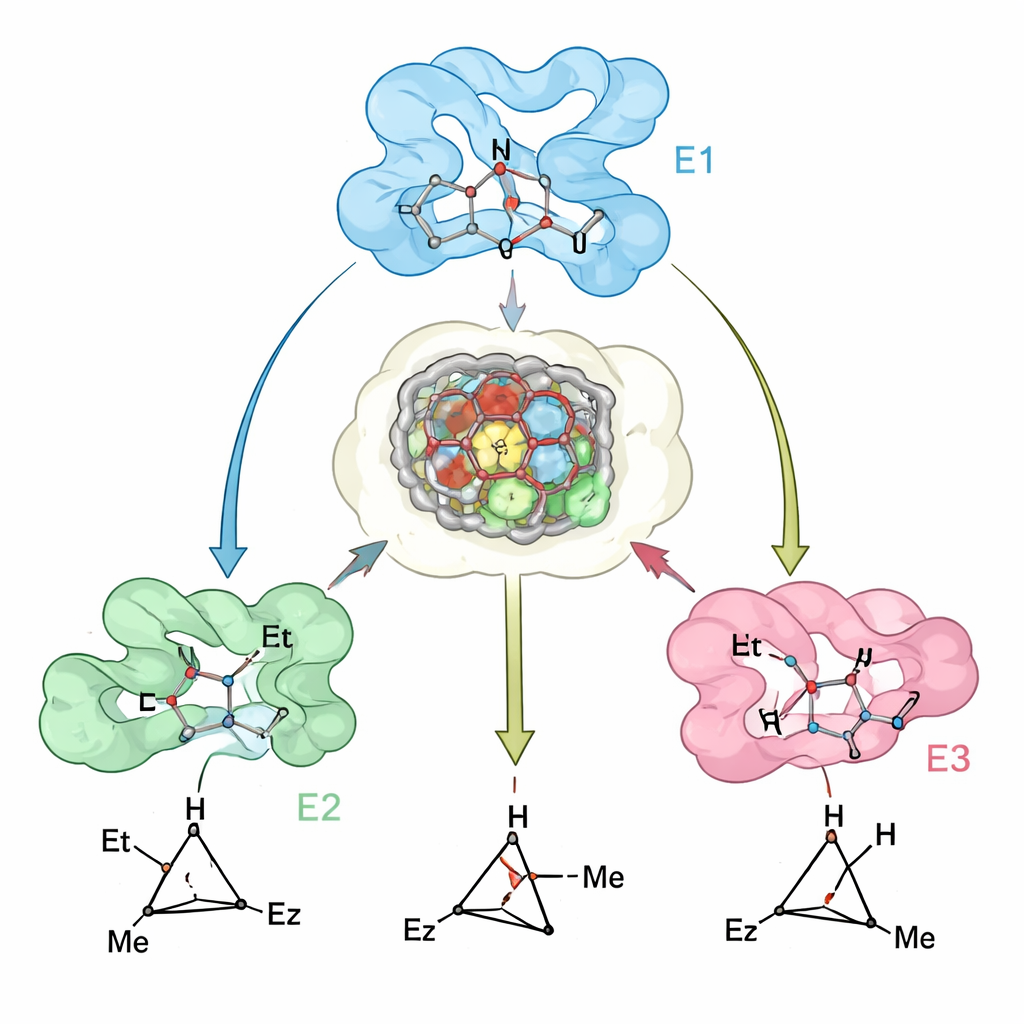
Wie gut das Design der Realität entspricht
Um zu prüfen, wie gut ihre Rechenmodelle reale Enzyme widerspiegeln, lösten die Forschenden Kristallstrukturen einer Schlüsselvariante von Myoglobin und verglichen sie mit den vorhergesagten Strukturen. Die Übereinstimmung war eng, und die experimentellen Daten hoben ein subtileres, aber wichtiges Merkmal hervor: Das Proteinzylinder‑Zentrum ist vororganisiert, um den bevorzugten Übergangszustand aufzunehmen, während kleine Verschiebungen in benachbarten Schleifen und Helices die Bindung des „falschen“ Übergangszustands energetisch ungünstig machen. Wo die Vorhersagen weniger genau waren — etwa bei einigen voluminösen Substraten — konnten die Abweichungen auf Rückgratbewegungen zurückgeführt werden, die in den Modellen nicht vollständig erfasst wurden, was klare Ansätze zur Verbesserung künftiger Designmethoden nahelegt.
Was das für zukünftige Medikamente und Katalysatoren bedeutet
Durch die Kombination physikbasierter Übergangszustandsmodellierung mit gezieltem Proteinumgestalten zeigt diese Arbeit, dass stereochemische Ergebnisse enzymkatalysierter Reaktionen vorab programmiert werden können, statt nur durch Versuch‑und‑Irrtum‑Evolution entdeckt zu werden. Die entstandene Palette an Cyclopropanasen bietet Chemikern einen praktischen Weg, vollständige Sätze von Cyclopropan‑Stereoisomeren aus einer breiten Palette von Ausgangs‑Olefinen herzustellen und vereinfacht dadurch Struktur‑Wirkungs‑Studien in der Arzneimittelentdeckung und bei der Synthese naturproduktähnlicher Moleküle erheblich. Dieselbe Strategie sollte sich auf andere Enzymtypen und Reaktionsklassen übertragen lassen und die Entwicklung von Biokatalysatoren beschleunigen, die präzise 3D‑Kontrolle über komplexe Moleküle liefern.
Zitation: Shen, Z., Siriboe, M.G., Ren, X. et al. Computational design of generalist cyclopropanases with stereodivergent selectivity. Nat Commun 17, 1620 (2026). https://doi.org/10.1038/s41467-026-68327-1
Schlüsselwörter: Biokatalyse, Cyclopropanierung, Enzymdesign, Stereochemie, Häm‑Proteine