Clear Sky Science · de
De-novo-Varianten im Spleißfaktor-Gen SF3B1 stehen im Zusammenhang mit neurodevelopmentalen Störungen
Wenn ein Gen die frühe Bauanleitung des Gehirns stört
Warum entwickeln einige Kinder Lernschwierigkeiten, Anfälle oder Fütterungsprobleme, obwohl Schwangerschaft und Geburt unauffällig erscheinen? Diese Studie untersucht ein einzelnes Gen namens SF3B1, das Zellen dabei hilft, genetische Botschaften zu verarbeiten. Die Forschenden zeigen, dass neue, spontane Veränderungen in diesem Gen die Art und Weise, wie Gehirnzellen DNA‑Anweisungen lesen, subtil durcheinanderbringen können und so ein bislang nicht erkanntes neurodevelopmentales Syndrom verursachen.
Ein zentraler Redakteur genetischer Botschaften
Jede Zelle unseres Körpers muss den rohen genetischen Text in klare Anweisungen umwandeln, bevor sie Proteine herstellen kann. Dieser Bearbeitungsschritt, bekannt als RNA‑Spleißen, entfernt nichtkodierende Abschnitte und fügt die brauchbaren Stücke zusammen. SF3B1 ist ein zentraler Bestandteil der zellulären „Spleißmaschine“. Bisher waren Veränderungen in SF3B1 vor allem aus dem Krebsbereich bekannt, wo Tumorzellen im Laufe des Lebens Mutationen in diesem Gen erwerben. Die neue Arbeit stellt eine andere Frage: Was passiert, wenn eine schädigende Veränderung in SF3B1 von der Empfängnis an in jeder Körperzelle vorhanden ist?
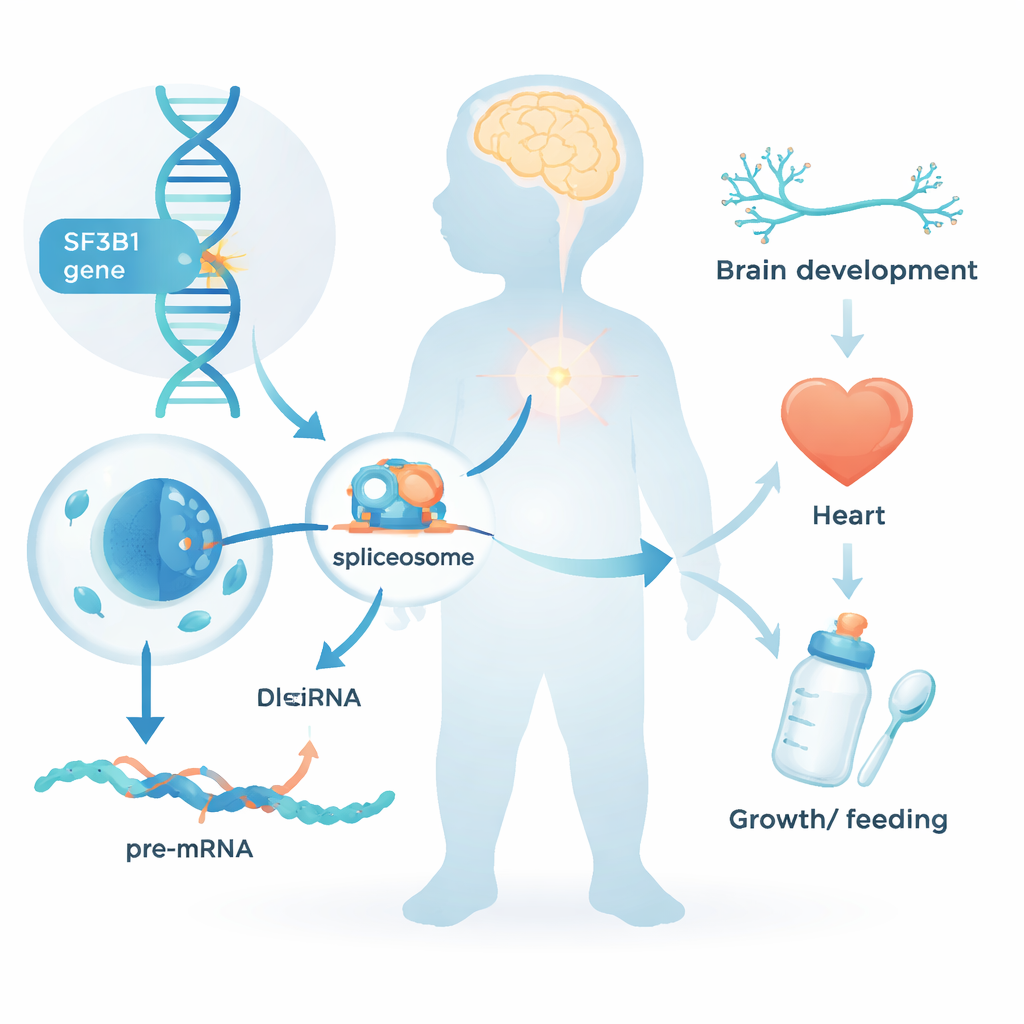
Ein neu erkanntes Kindheitssyndrom
Das Team sammelte Daten von 26 Kindern und jungen Erwachsenen, die alle seltene SF3B1‑Varianten trugen, meist de novo entstanden — das heißt, nicht von einem der Eltern vererbt. Fast alle zeigten neurodevelopmentale Probleme: Verzögerungen beim Sitzen, Gehen oder Sprechen; eine intellektuelle Beeinträchtigung überwiegend von leichter bis mittelschwerer Ausprägung; und bei etwa der Hälfte der Fälle Anfälle. Viele hatten niedrigen Muskeltonus und benötigten zusätzliche Unterstützung beim Füttern, manchmal über eine Magensonde. Die Gesichtszüge waren subtil untypisch, jedoch nicht bei jedem Kind gleich; ein auffälliges, häufiges Merkmal war ein hoher oder gespaltenes Gaumen. Mehrere Teilnehmende wiesen zudem Herzfehler, Wachstumsrestriktion oder einen kleinen Kopfumfang auf, was zeigt, dass die Auswirkungen von SF3B1‑Veränderungen über das Gehirn hinausgehen.
Zwei Klassen genetischer Veränderungen, zwei klinische Muster
Die Forschenden unterschieden zwischen zwei großen Typen von SF3B1‑Varianten. Zur einen Gruppe gehörten „Loss‑of‑Function“-Veränderungen, etwa vorzeitige Stoppcodons, die voraussichtlich die Menge an funktionsfähigem SF3B1‑Protein verringern. Die zweite Gruppe umfasste Missense‑Varianten, bei denen eine einzelne Aminosäure im Protein ausgetauscht ist. Durch das Clustern der medizinischen Merkmale der Kinder beobachtete das Team, dass Träger von Missense‑Varianten tendenziell schwerere und komplexere Probleme hatten, einschließlich höherer Raten von Herz‑ und Magen‑Darm‑Anomalien, Kleinwuchs und Mikrozephalie. Loss‑of‑Function‑Varianten dagegen wurden manchmal von einem leicht betroffenen oder sogar scheinbar gesunden Elternteil vererbt, was darauf hindeutet, dass eine einfach verminderte SF3B1‑Menge bei manchen Personen mit relativ milden Symptomen vereinbar sein kann.
Feinabstimmungsfehler statt kompletter Zusammenbruch
Um zu verstehen, was die Missense‑Varianten in Zellen bewirken, rekonstruierten die Wissenschaftler sie in Laborzelllinien. Überraschenderweise konnten diese veränderten SF3B1‑Proteine die grundlegende Spleißfunktion noch ausreichend erfüllen, um Zellen zu retten, in denen das normale SF3B1 ausgeschaltet worden war. Das schloss eine einfache Loss‑of‑Function‑Erklärung aus. Mithilfe tiefer RNA‑Sequenzierung betrachtete das Team dann das gesamte Spektrum zellulärer Botschaften. Sie fanden, dass Missense‑Varianten das Spleißen von Hunderten Genen subtil verschoben — insbesondere durch die Veränderung der Auswahl von Spleißstellen an den Enden von Exons und durch gelegentliches Exon‑Skipping. Das Ausmaß der Störung war geringer als bei der klassischen krebsassoziierten SF3B1‑Mutation K700E, aber dennoch erheblich: Viele betroffene Gene sind an der Gehirnentwicklung, der Vernetzung von Nerven sowie an grundlegenden Prozessen wie RNA‑Verarbeitung und Proteinsynthese beteiligt.
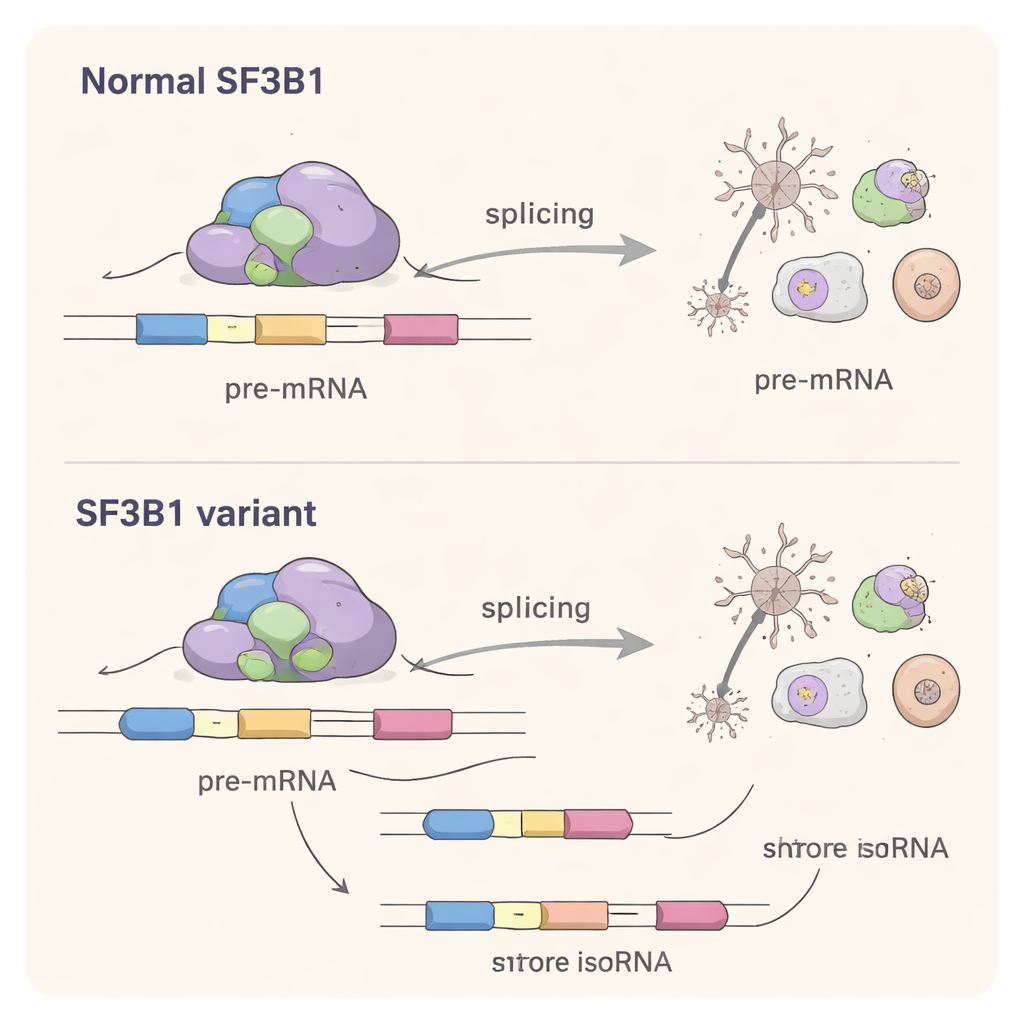
Ein geteilter Mechanismus zwischen Krebs und Hirnerkrankungen
Obwohl die meisten neurodevelopmentalen SF3B1‑Varianten an anderen Positionen auftreten als die bekannten Krebsmutationen, stören sie denselben Kernprozess: die präzise Erkennung von Spleißstellen in der RNA. Die Studie zeigt, dass diese Entwicklungsvarianten ein eigenes „Spleiß‑Signaturmuster“ besitzen und alternative Spleißstellen wählen, die häufig näher an den normalen liegen als jene, die im Krebs bevorzugt werden. Das spricht für einen Change‑of‑Function‑Mechanismus, bei dem das mutierte Protein mit der normalen Kopie konkurriert und die Spleißmaschine dazu bringt, bei vielen Genen gleichzeitig leicht fehlerhafte Entscheidungen zu treffen.
Was das für Familien und zukünftige Forschung bedeutet
Für betroffene Familien identifiziert die Arbeit SF3B1 als neue Ursache neurodevelopmentaler Störungen, die nun in genetischen Kliniken getestet werden kann und damit langwierige Diagnosesuchen beenden könnte. Allgemeiner erweitert sie SF3B1 die kleine, aber wachsende Liste von Spleißgenen, deren Veränderungen je nach Zeitpunkt und Art der Veränderung sowohl Krebs als auch kindliche Hirnstörungen antreiben können. Indem die Studie kartiert, wie spezifische SF3B1‑Varianten das RNA‑Spleißen umgestalten, legt sie die Grundlage für künftige Therapien, die darauf abzielen, Fehl‑Spleißen gezielt zu korrigieren.
Zitation: Uguen, K., Bergot, T., Scott-Boyer, MP. et al. De novo variants in the splicing factor gene SF3B1 are associated with neurodevelopmental disorders. Nat Commun 17, 1569 (2026). https://doi.org/10.1038/s41467-026-68284-9
Schlüsselwörter: RNA-Spleißen, SF3B1, neurodevelopmentale Störungen, De‑novo‑Varianten, Spliceosomopathien