Clear Sky Science · de
Reduktion von RAD23A verlängert die Lebensspanne und mildert Pathologie in einem Mausmodell der TDP-43-Proteinopathie
Warum diese Forschung für Familien und Patientinnen/Patienten wichtig ist
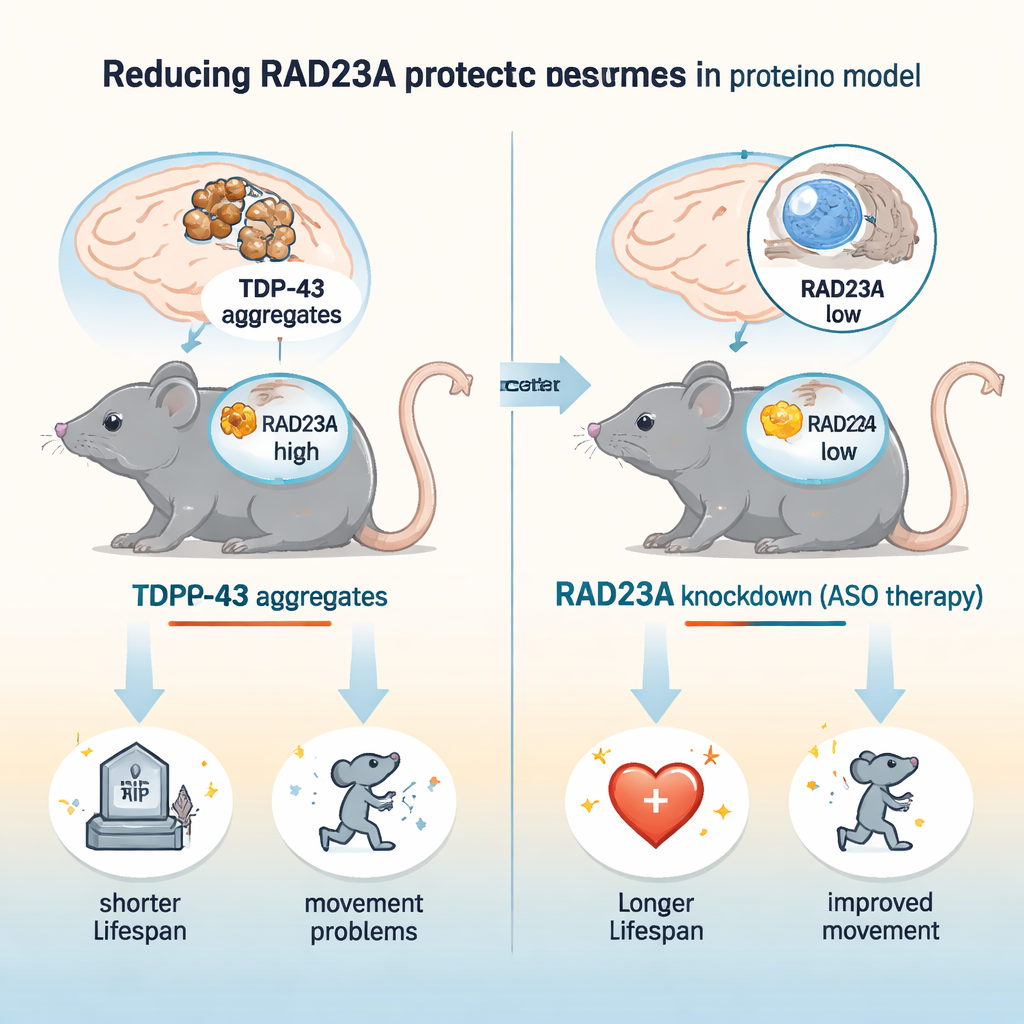
Viele Formen von Demenz und Motoneuronerkrankungen, darunter amyotrophe Lateralsklerose (ALS) und frontotemporale Demenz (FTD), sind durch Proteine in Nervenzellen gekennzeichnet, die falsch gefaltet sind, verklumpen und die Neurone über die Zeit vergiften. Einer der Hauptschuldigen ist das Protein TDP-43, das normalerweise bei der RNA-Verwaltung hilft, bei Aggregation jedoch toxisch wird. Die Studie stellt eine hoffnungsvolle Frage: Können wir Nervenzellen widerstandsfähiger machen, indem wir ein anderes Protein namens RAD23A drosseln, das an der Handhabung beschädigter Proteine beteiligt ist? Die Autorinnen und Autoren zeigen an Mäusen, dass die Verminderung von RAD23A das Leben verlängern, die Bewegungsfähigkeit verbessern und die Hirnschädigung in einem Modell für TDP-43-getriebene Erkrankung verringern kann – ein Hinweis auf eine mögliche neue Behandlungsstrategie.
Ein Proteinstau in kranken Neuronen
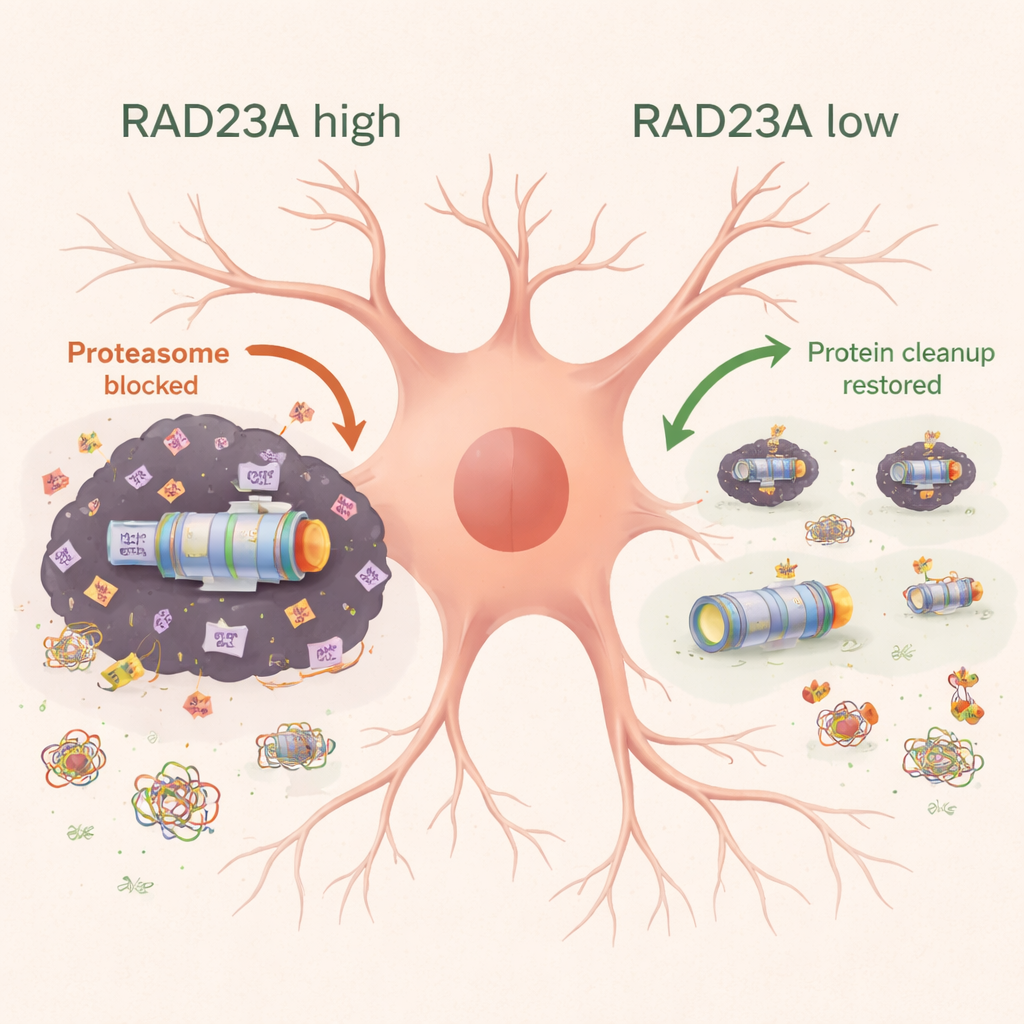
Neurodegenerative Erkrankungen zeigen häufig Haufen fehlgefalteter Proteine, die von den zellulären Abfallentsorgungsmechanismen nicht entfernt werden können. Bei ALS und FTD verlässt TDP-43 den Zellkern, bildet klebrige Aggregate und wird stark mit Ubiquitin markiert – einem Signal, das normalerweise Proteine zum Proteasom, der zellulären Schreddermaschine, dirigiert. RAD23A ist eines von mehreren „Shuttle“-Proteinen, die ubiquitinmarkierte Ladung zum Proteasom transportieren können. Frühere Arbeiten in Würmern und kultivierten Neuronen deuteten jedoch darauf hin, dass der Verlust von RAD23-ähnlichen Proteinen paradoxerweise vor TDP-43–induzierten Schäden schützen könnte. Diese Studie untersuchte dieses Paradoxon im lebenden Säugerhirn.
RAD23A herunterregulieren in einem TDP-43-Mausmodell
Die Forschenden verwendeten ein etabliertes Mausmodell, genannt TAR4/4, das menschliches TDP-43 in Neuronen überexprimiert und Bewegungsprobleme, Wirbelsäulenverkrümmung, Tremor und vorzeitigen Tod entwickelt – Merkmale, die ALS/FTD ähneln. Sie reduzierten RAD23A auf zwei Wegen: durch Injektion von Neugeborenen mit Antisense-Oligonukleotiden (ASOs), die Rad23a-RNA vermindern, und durch Zucht von Mäusen mit einem genetischen Rad23a-Knockout. Eine einzelne ASO-Behandlung senkte die RAD23A-Spiegel im Gehirn und Rückenmark um etwa drei Viertel. In diesen TDP-43-Mäusen verlängerte die RAD23A-Reduktion die Lebensspanne um ungefähr 50 % und verzögerte Beginn und Schwere von Gangstörungen, Tremor, Wirbelsäulenverkrümmung und Hinterbein-Klammern. Interessanterweise brachte ein kompletter genetischer Verlust von RAD23A keinen zusätzlichen Nutzen, was darauf hindeutet, dass eine partielle Reduktion optimal ist und ein langfristiges vollständiges Fehlen kompensatorische Veränderungen auslösen könnte.
Weniger Entzündung, sauberere Proteinhomöostase und ein ruhigeres Genom
Mikroskopische Untersuchungen des Motorkortex zeigten, dass TDP-43-Mäuse Neurone verloren und eine starke Aktivierung von Astrozyten und Mikroglia – den Stütz- und Immunzellen des Gehirns – entwickelten. Die Senkung von RAD23A bewahrte die Anzahl der Neurone und reduzierte Marker für Entzündung und Zelltod. Biochemische Analysen zeigten, dass TDP-43-Überproduktion die Zellen mit ubiquitinmarkierten, detergentin- solubilisierungsresistenten Proteinen überflutete und Proteasomuntereinheiten in diese Aggregate zog, wodurch die Fähigkeit der Zelle, beschädigte Proteine zu beseitigen, geschwächt wurde. Die Reduktion von RAD23A senkte die Gesamtlast ubiquitinierter Proteine, hielt mehr Proteasome in der löslichen, funktionalen Pool und stellte mehrere Arten von Proteasomaktivität annähernd wieder her. Gleichzeitig verringerte die RAD23A-Reduktion sowohl die Gesamt- als auch die aggregierten Formen von TDP-43, einschließlich eines besonders toxischen 25-Kilodalton-Fragments, und verlagerte TDP-43 aus dem Zytoplasma zurück in den Zellkern. Genomweite RNA-Sequenzierung zeigte, dass Tausende von durch TDP-43 ausgelösten Genexpressionsänderungen teilweise rückgängig gemacht wurden, insbesondere Gene, die an neuronaler Funktion, mitochondrialer Energieproduktion und aggregate-abbauenden Wegen wie der Aggrephagie beteiligt sind.
Umgestaltung des versteckten „unlöslichen“ Proteoms
Um die hartnäckigen Aggregate, die normalen Detergentien widerstehen, genauer zu untersuchen, nutzte das Team Schwerisotopen-Massenspektrometrie, um Proteine zu katalogisieren, die in der unlöslichen Fraktion der Mauscortex gefangen sind. Die Expression von menschlichem TDP-43 zog Proteasomkomponenten, Zytoskelett- und Transportproteine sowie andere zelluläre Maschinerie an. Als RAD23A herunterreguliert wurde, verschob sich die Gesamtzusammensetzung dieses unlöslichen Proteoms: Weniger proteasom- und transportbezogene Proteine wurden sequestriert, während einige ribosomale und stressbezogene Proteine in den Aggregaten zunahmen. Bemerkenswerterweise spiegelte diese Umgestaltung nicht einfach Änderungen auf RNA-Ebene wider, was darauf hindeutet, dass RAD23A vornehmlich beeinflusst, wie vorhandene Proteine zwischen löslichen und aggregierten Zuständen verteilt sind, und weniger, wie viel von jedem Protein synthetisiert wird.
Was das für künftige Therapien bedeuten könnte
Zusammen zeichnen diese Befunde ein Bild von RAD23A als starken Regulator der Protequalitätskontrolle in gestressten Neuronen. Durch partielle Senkung von RAD23A in einem TDP-43–getriebenen Mausmodell konnten die Autorinnen und Autoren toxische Proteinaggregate reduzieren, die Aktivität der Proteinentsorgungsmaschinerie wiederherstellen, schädliche Genexpressionsänderungen dämpfen, Hirnentzündung begrenzen und Leben sowie motorische Funktionen verlängern. Da abnorme TDP-43-Akkumulation sowohl in erblichen als auch in sporadischen Formen von ALS, FTD und verwandten Störungen weit verbreitet ist, könnte die Zielsteuerung von RAD23A mit für Menschen geeigneten Antisense-Wirkstoffen einen Weg bieten, Neurone zu schützen, ohne TDP-43 direkt zu blockieren, das eine essentielle Funktion erfüllt. Obwohl noch viel in anderen Modellen und beim Menschen geprüft werden muss, identifiziert diese Arbeit RAD23A als vielversprechenden neuen Ansatzpunkt für einen gemeinsamen Weg der Neurodegeneration.
Zitation: Guo, X., Prajapati, R.S., Chun, J. et al. Reduction of RAD23A extends lifespan and mitigates pathology in a mouse model of TDP-43 proteinopathy. Nat Commun 17, 1820 (2026). https://doi.org/10.1038/s41467-025-65104-4
Schlüsselwörter: TDP-43, ALS, Proteinaggregation, Proteasom, Antisense-Therapie