Clear Sky Science · de
Beste Praktiken und Werkzeuge in R und Python für die statistische Verarbeitung und Visualisierung von Lipidom- und Metabolomikdaten
Warum es wichtig ist, Laborzahlen in klare Bilder zu verwandeln
Moderne Messgeräte können inzwischen tausende winziger Moleküle – Lipide und andere Metaboliten – in einem einzigen Tropfen Blut oder Gewebe messen. Diese Messungen enthalten Hinweise auf Krankheitsrisiken, Therapieansprechen und darauf, wie unser Körper auf Ernährung oder Alter reagiert. Die Rohdaten sind jedoch keine fertige Antwort: sie bestehen aus großen Zahlentabellen, die bereinigt, analysiert und in verständliche Darstellungen überführt werden müssen. Dieser Artikel erklärt, wie Forschende zwei gängige Programmiersprachen, R und Python, nutzen können, um dies zuverlässig, transparent und mit publikationsreifen Grafiken zu tun.
Von chemischen Messungen zu komplexen Datentabellen
In der Lipidomik und Metabolomik erzeugen Massenspektrometrie und Chromatographie große Datensätze, bei denen jede Zeile eine Probe und jede Spalte ein Molekül darstellt. Diese Tabellen verhalten sich selten wie ordentliche Lehrbuchbeispiele. Sie enthalten fehlende Werte, Ausreißer und verzerrte Verteilungen, bei denen einige wenige Moleküle extrem hohe Werte zeigen. Konzentrationen können mehrere Größenordnungen umfassen und von Alter, Geschlecht, Ernährung, Medikamenten, tageszeitlichen Schwankungen oder technischen Problemen wie Instrumentdrift oder Chargeneffekten beeinflusst sein. Internationale Expertengruppen haben Richtlinien zur Standardisierung der Probenentnahme, -verarbeitung und -berichterstattung herausgegeben, aber selbst bei guter Laborpraxis bleibt eine sorgfältige statistische Aufbereitung notwendig, um wahre biologische Signale aus diesem verrauschten Hintergrund zu extrahieren.
Bereinigen und Vorbereiten der Zahlen
Bevor ein Vergleich zwischen gesunden und erkrankten Gruppen sinnvoll ist, müssen die Daten vorbereitet werden. Die Übersichtsarbeit beschreibt, wie fehlende Werte entstehen – durch zufällige Pannen, Messgrenzen des Instruments oder Signalstörungen – und erklärt, wann sie vernachlässigt werden können, wann nachgemessen werden sollte und wie sie sinnvoll geschätzt (imputiert) werden können, etwa mit k-nächsten Nachbarn, Random Forests oder einfacher Substitution durch niedrige Werte. Anschließend skizzieren die Autor:innen Normalisierungsstrategien, die unerwünschte Variationen reduzieren, zum Beispiel durch Korrektur von Chargeneffekten mit Qualitätskontrollproben oder durch Anpassung an Unterschiede in der Probenmenge. Danach werden Transformationen wie Logarithmierungen diskutiert – die lange rechtsseitige Schwänze in den Daten zähmen – sowie Skalierungsmethoden, die alle Moleküle vergleichbar machen, damit stark variable Verbindungen spätere Analysen nicht dominieren. 
Statistische Tests und visuelle Erzählungen
Sind die Daten korrekt aufbereitet, kommen verschiedene statistische Werkzeuge zum Einsatz. Für einzelne Moleküle können Forschende Fold-Changes berechnen und klassische Tests wie den t-Test oder dessen nichtparametrische Gegenstücke (etwa den Mann–Whitney-Test) nutzen, um zu prüfen, ob sich die Konzentrationen zwischen Gruppen unterscheiden. Für Vergleiche mit mehreren Gruppen werden Methoden wie ANOVA oder der Kruskal–Wallis-Test eingeführt, begleitet von Post-hoc-Verfahren, um zu bestimmen, welche Gruppen sich unterscheiden. Die Aussagekraft dieser Tests entfaltet sich, wenn ihre Ergebnisse klar visualisiert werden. Der Artikel hebt Boxplots (einschließlich verbesserter Versionen für schiefe Daten), Violinplots und Volcano-Plots hervor, die Effektgröße und statistische Signifikanz kombinieren. Für Lipide werden spezialisiertere Visualisierungen beschrieben, etwa Lipidnetzwerke, die koordinierte Veränderungen ganzer Klassen zeigen, sowie Darstellungen der Fettsäureketten, die Muster in Kohlenstoffkettenlänge und Sättigung aufdecken.
Mustererkennung bei vielen Variablen gleichzeitig
Da jede Probe Hunderte oder Tausende gemessener Moleküle enthalten kann, sind multivariate Methoden entscheidend. Die Übersichtsarbeit erklärt, wie die Hauptkomponentenanalyse (PCA) diese Komplexität auf wenige neue Achsen komprimiert, die die wichtigsten Richtungen der Variation erfassen und schnelle Kontrollen auf Gruppenunterscheidung, Chargeneffekte oder analytische Stabilität erlauben. Fortgeschrittene nichtlineare Methoden wie t-SNE und UMAP können subtile Cluster und Strukturen im hochdimensionalen Raum sichtbar machen. Für Situationen, in denen das Ziel die Klassifikation von Proben ist – z. B. Unterscheidung von Patient:innen und Kontrollen – beschreiben die Autor:innen überwachte Ansätze auf Basis von Partial Least Squares und dessen orthogonaler Erweiterung (PLS-DA und OPLS-DA). Diese Methoden verknüpfen molekulare Profile mit Probenlabels, unterstützen Feature-Auswahl und werden häufig mit Scoreplots, Loadingplots und ROC-Kurven zusammengefasst.
Praktische Toolkits in R und Python
Um Einsteiger:innen den Übergang von Theorie zu Praxis zu erleichtern, stellt der Artikel ein breites Ökosystem an Softwarepaketen vor. In R vereinfachen Sammlungen wie tidyverse und tidymodels Datenaufbereitung und Modellierung, während ggplot2 und Zusatzpakete wie ggpubr, ggstatsplot und tidyplots das Erstellen publikationsreifer Grafiken erleichtern. Spezialisierte Bibliotheken behandeln PCA, Clustering und PLS-basierte Modelle, und Bioconductor-Pakete unterstützen komplexe Heatmaps und interaktive Grafiken. In Python übernimmt pandas die Tabellenverarbeitung, während matplotlib, seaborn und plotly die Visualisierung abdecken und scikit-learn eine breite Palette multivariater Methoden bietet. Die Autor:innen betonen durchgehend schrittweise Beispiele, die in einem begleitenden GitBook verfügbar sind, sodass Leser:innen Workflows reproduzieren und an ihre eigenen Daten anpassen können. 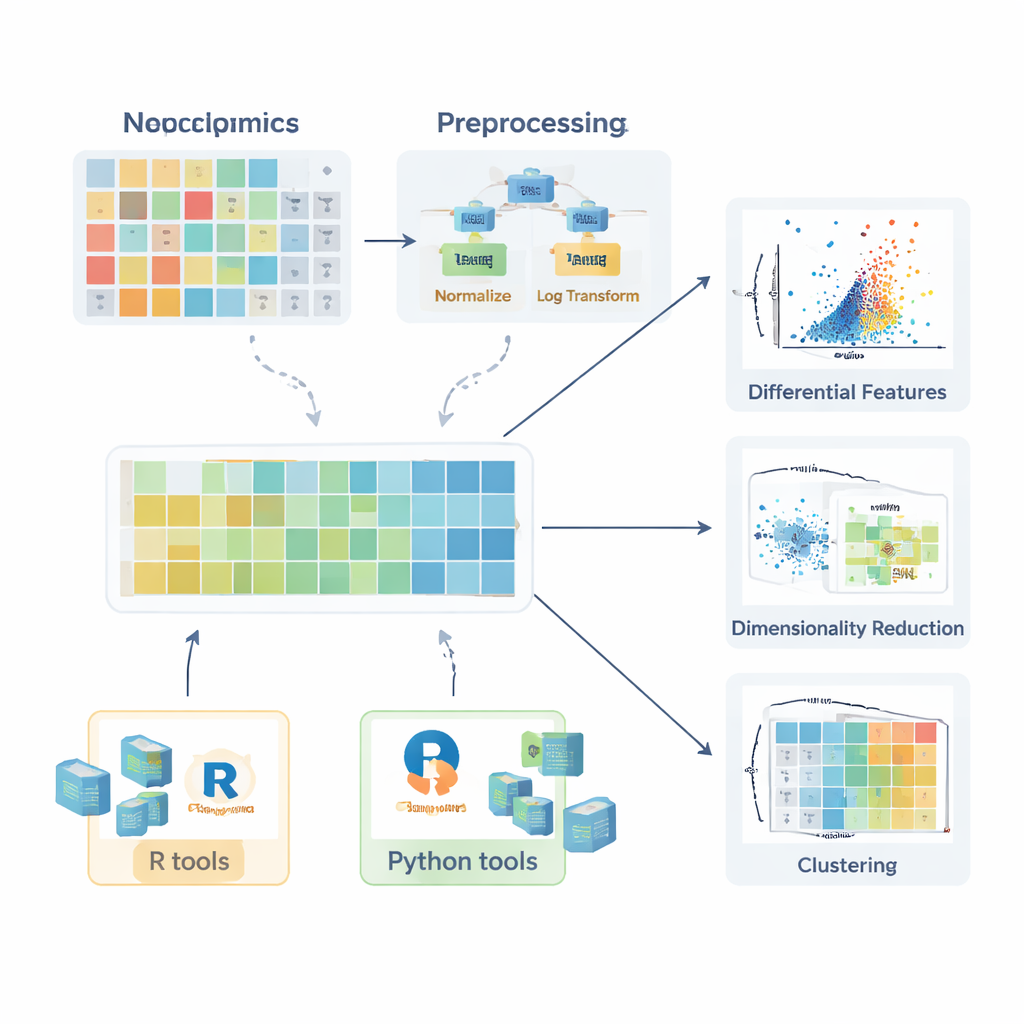
Komplexe Chemie in verlässliche Einsichten verwandeln
Der Artikel schließt mit der Feststellung, dass das eigentliche Potenzial der Lipidomik und Metabolomik nicht nur in leistungsfähigen Geräten liegt, sondern darin, wie durchdacht deren Ausgabe verarbeitet und visualisiert wird. Durch Befolgung guter statistischer Praxis, die Nutzung offener und gut dokumentierter Werkzeuge in R und Python und das Vertrauen auf gemeinsam genutzte Codebeispiele können Forschende robuste und reproduzierbare Pipelines aufbauen. Das erhöht die Wahrscheinlichkeit, dass in winzigen Molekülen gefundene Muster in verlässliche Biomarker, verbessertes Verständnis von Krankheitsmechanismen und personalisiertere medizinische Ansätze münden, die letztlich Patientinnen und Patienten zugutekommen.
Zitation: Idkowiak, J., Dehairs, J., Schwarzerová, J. et al. Best practices and tools in R and Python for statistical processing and visualization of lipidomics and metabolomics data. Nat Commun 16, 8714 (2025). https://doi.org/10.1038/s41467-025-63751-1
Schlüsselwörter: Lipidomik, Metabolomik, Datenvisualisierung, R-Programmierung, Python