Clear Sky Science · de
Kombination von räumlicher Transkriptomik mit Gewebemorphologie
Blicke ins Gewebe auf zweifache Weise
Ärztinnen, Ärzte und Wissenschaftler möchten zunehmend nicht nur wissen, welche Gene in einem Gewebe aktiv sind, sondern genau, an welchen Orten sie angeschaltet werden. Gleichzeitig liefern Krankenhausmikroskope bereits reichhaltige Bilder der Gewebestruktur, die Pathologinnen und Pathologen täglich nutzen. Dieser Artikel erklärt, wie ein neues Forschungsfeld versucht, diese beiden Sichtweisen — detaillierte Karten der Genaktivität und gewöhnliche Mikroskopaufnahmen — miteinander zu verbinden, und warum diese Verbindung zu früheren Diagnosen, besserer Krebsgrading und tieferem Verständnis davon führen könnte, wie Krankheiten entstehen und sich ausbreiten.
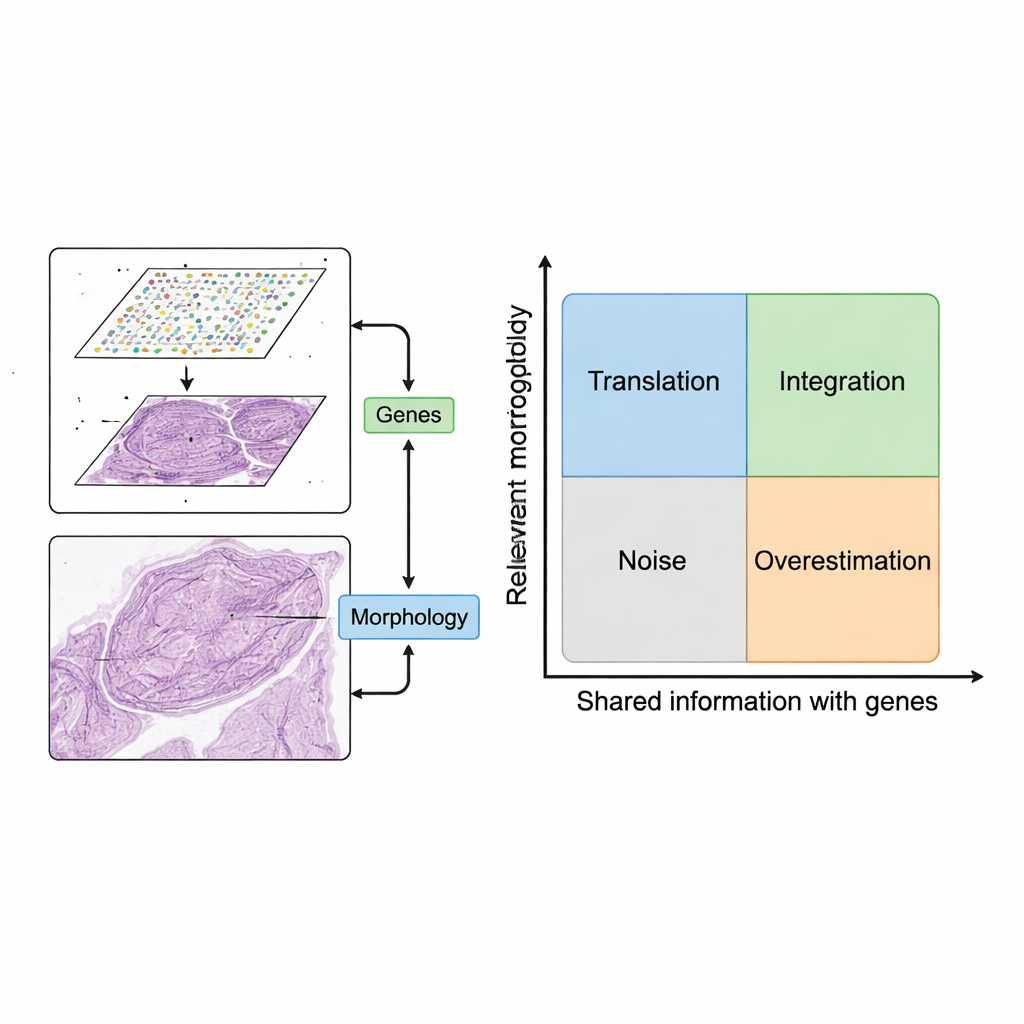
Von verstreuten Zellen zu Karten der Genaktivität
Jahrelang erforderten leistungsfähige „-omics“-Methoden, dass Gewebe zu einer Mischung aus Einzelzellen zerkleinert werden, wodurch Informationen darüber verloren gingen, wo sich jede Zelle im Gewebe befand. Die räumliche Transkriptomik änderte dies, indem sie die Genaktivität misst und zugleich die Position jeder Zelle im Gewebe beibehält. Das Ergebnis ist ein Raster von Messpunkten, jeweils mit einem Genexpressionsprofil und präzisen Koordinaten. Alleinstehend hat diese räumliche Geninformation bereits neue Muster zellulärer Vielfalt und Krankheitsarchitekturen aufgedeckt. Sie ist jedoch unveränderlich, sobald sie gemessen wurde, und das Experiment zu wiederholen ist teuer. Im Gegensatz dazu sind mit Standardfärbungen wie der weit verbreiteten Hämatoxylin‑Eosin (H&E)-Färbung angefertigte Gewebebilder günstig, häufig vorhanden und enthalten visuelle Hinweise auf Zellform, Dichte und Gewebeorganisation.
Zwei Wege, Bilder und Gene zu kombinieren
Die Übersichtsarbeit schlägt einen einfachen, aber wirkungsvollen Rahmen vor, um diese beiden Datenquellen zusammenzuführen. Zuerst werden Bildausschnitte mit nahegelegenen Genexpressionspunkten gepaart. Dann extrahieren Computer‑Modelle Merkmale aus den Bildern — Muster, die Form, Textur und Organisation erfassen — und vergleichen sie mit Mustern in der Genexpression. Die Autoren beschreiben zwei erwünschte Szenarien. Bei der „Translation“ folgen Bildmerkmale eng relevanter Genaktivität, sodass Modelle mithilfe nur des Gewebebilds vorhersagen können, welche Gene aktiv sind. Das lässt sich nutzen, um fehlende Genmessungen zu ergänzen, eine feinere Auflösung als das ursprüngliche Raster zu erreichen oder Genaktivität aus routinemäßigen klinischen Schnitten zu erschließen, ohne zusätzliche Laborarbeit. Bei der „Integration“ hingegen erfassen Bildmerkmale nützliche Informationen, die die Gen‑Daten übersehen, etwa langsame strukturelle Veränderungen oder subtile Gewebeorganisation, und helfen so, klarere Regionen oder „Domänen“ innerhalb eines Gewebes zu definieren.
Wenn zusätzliche Information hilft — und wann sie schadet
Nicht jedes Bildmerkmal ist nützlich. Die Autoren führen eine konzeptionelle Karte mit zwei Achsen ein: wie relevant ein Bildmerkmal für die biologische Fragestellung ist und wie stark es mit Geninformationen überlappt. Merkmale, die weder relevant noch mit Genen verwandt sind, sind bloßes Rauschen, etwa Färbeartefakte. Merkmale, die Genmuster nachzeichnen, aber an unbedeutende Gene gebunden sind (wie grundlegende „Housekeeping“-Gene), können Modelle auf dem Papier gut aussehen lassen, aber wenig klinischen Wert hinzufügen. Durch die Einordnung von Methoden in vier Quadranten — Translation, Integration, Rauschen und Überschätzung — macht der Rahmen deutlich, wann das Kombinieren von Bildern und Genen wirklich neue Einsichten bringt und wann es nur Bekanntes wiederholt oder verschleiert.
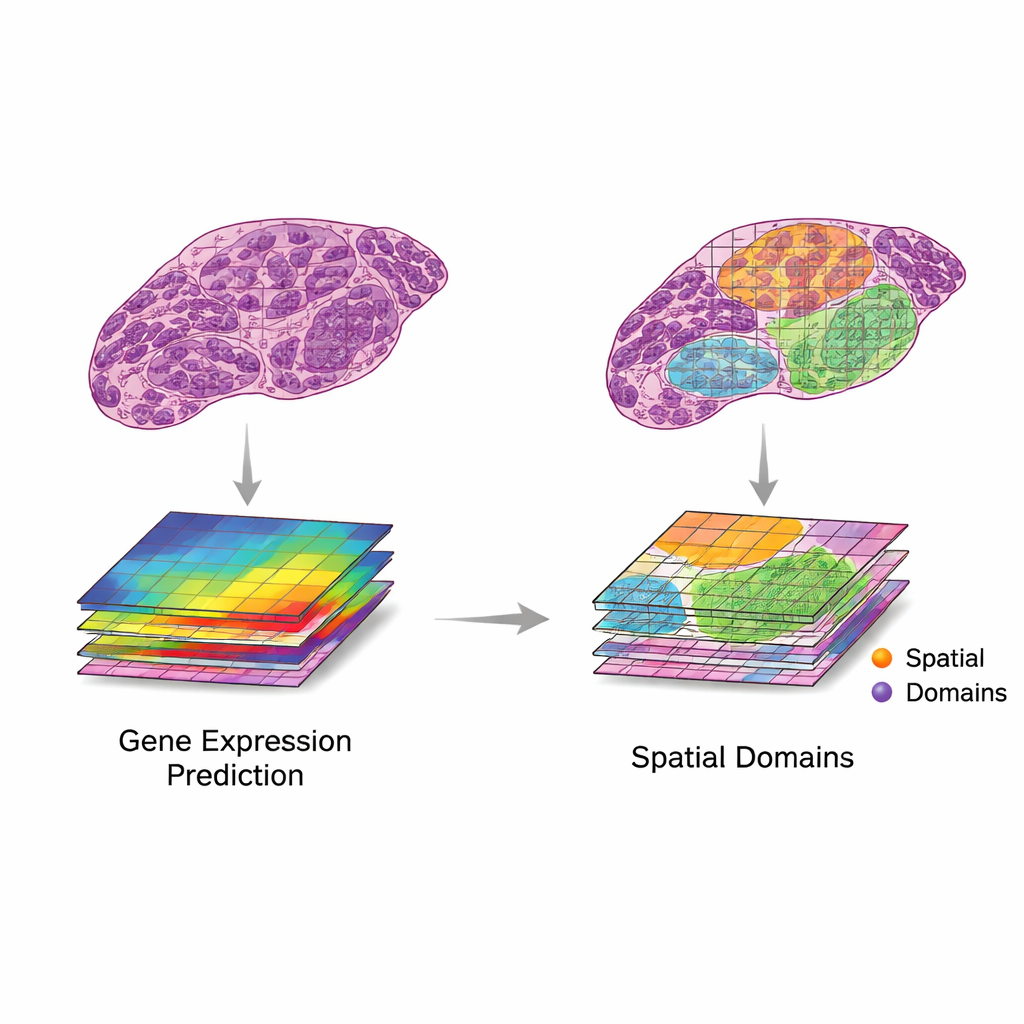
Aktuelle Werkzeuge, Tests und Wachstumsprobleme
Eine schnell voranschreitende Welle von Methoden der künstlichen Intelligenz versucht inzwischen, Translation und Integration an realen Daten durchzuführen. Frühe Systeme setzten auf konvolutionale neuronale Netze, während neuere Ansätze Transformer, Graph‑Neuronale Netze und Multi‑Skalen‑Modelle nutzen, die Details von winzigen Zellstrukturen bis zum gesamten Schnitthintergrund erfassen können. Diese Methoden wurden verwendet, um Genaktivität aus H&E‑Bildern vorherzusagen, Super‑Auflösungs‑Karten zu erzeugen und Geweberegionen mit unterschiedlichem Verhalten zu identifizieren. Zur Beurteilung der Leistung stützen sich Forschende auf statistische Maße wie die Korrelation zwischen vorhergesagten und beobachteten Genleveln oder die Übereinstimmung zwischen KI‑definierten Regionen und Expertenlabels von Pathologinnen und Pathologen. Die Datensätze sind jedoch noch klein und unterschiedlich, und Vergleiche über Studien hinweg sind schwierig. Viele berichtete Verbesserungen könnten Überanpassung widerspiegeln oder Erfolge bei Genen und Mustern zeigen, die klinisch wenig Bedeutung haben.
Wohin das führen könnte
Die Autoren kommen zu dem Schluss, dass die Kombination räumlicher Genkarten mit Gewebebildern vielversprechend, aber noch früh in ihrer Entwicklung ist. Die heutigen Modelle erreichen oft nur moderate Genauigkeit und sind bisher nicht für den routinemäßigen medizinischen Einsatz gewappnet. Künftige Fortschritte werden wahrscheinlich von besseren Bildmerkmalen ausgehen, insbesondere von großen „Foundation Models“, die auf Millionen pathologischer Schnitte trainiert wurden, und davon, den Fokus auf Gene und Muster zu legen, die die Patientenversorgung tatsächlich beeinflussen. Sorgfältig gestaltete Integration könnte eines Tages Frühwarnzeichen von Krankheiten offenbaren, indem sie Diskrepanzen zwischen dem aktuellen Erscheinungsbild des Gewebes und dem, was seine Gene voraussagen, erkennt. Kurz gesagt, diese Arbeit zeichnet eine Roadmap dafür, wie sich routinemäßige mikroskopische Bilder in reichhaltige, geninformierte Karten verwandeln lassen, die Ärztinnen und Ärzten helfen, Krankheiten präziser zu verstehen und zu behandeln.
Zitation: Chelebian, E., Avenel, C. & Wählby, C. Combining spatial transcriptomics with tissue morphology. Nat Commun 16, 4452 (2025). https://doi.org/10.1038/s41467-025-58989-8
Schlüsselwörter: räumliche Transkriptomik, Gewebemorphologie, digitale Pathologie, Vorhersage der Genexpression, Imaging-KI