Clear Sky Science · de
Loss-of-function-Varianten in ODAD1 stören ODA-Andocken und induzieren Remodellierung des Aktin-Zytoskeletts bei primärer ziliärer Dyskinesie
Wenn die mikroskopischen Bürsten des Körpers versagen
Jeder Atemzug hängt von Millionen winziger, haarähnlicher Strukturen namens Zilien ab, die Schleim, Keime und Staub aus den Atemwegen fegen. Bei Menschen mit einer seltenen erblichen Erkrankung, der primären ziliären Dyskinesie (PCD), funktionieren diese mikroskopischen Bürsten nicht richtig, was zu hartnäckigem Husten, Lungeninfektionen und manchmal zu einer umgekehrten Lage der inneren Organe führt. Diese Studie legt dar, wie schädliche Veränderungen in einem einzigen Gen, ODAD1, nicht nur die internen Motoren der Zilien außer Gefecht setzen, sondern überraschenderweise auch das Gerüst der Zellen, die die Zilien tragen, umgestalten — und damit einen neuen, potenziell medikamentös ansprechbaren Schwachpunkt der Krankheit offenbaren.
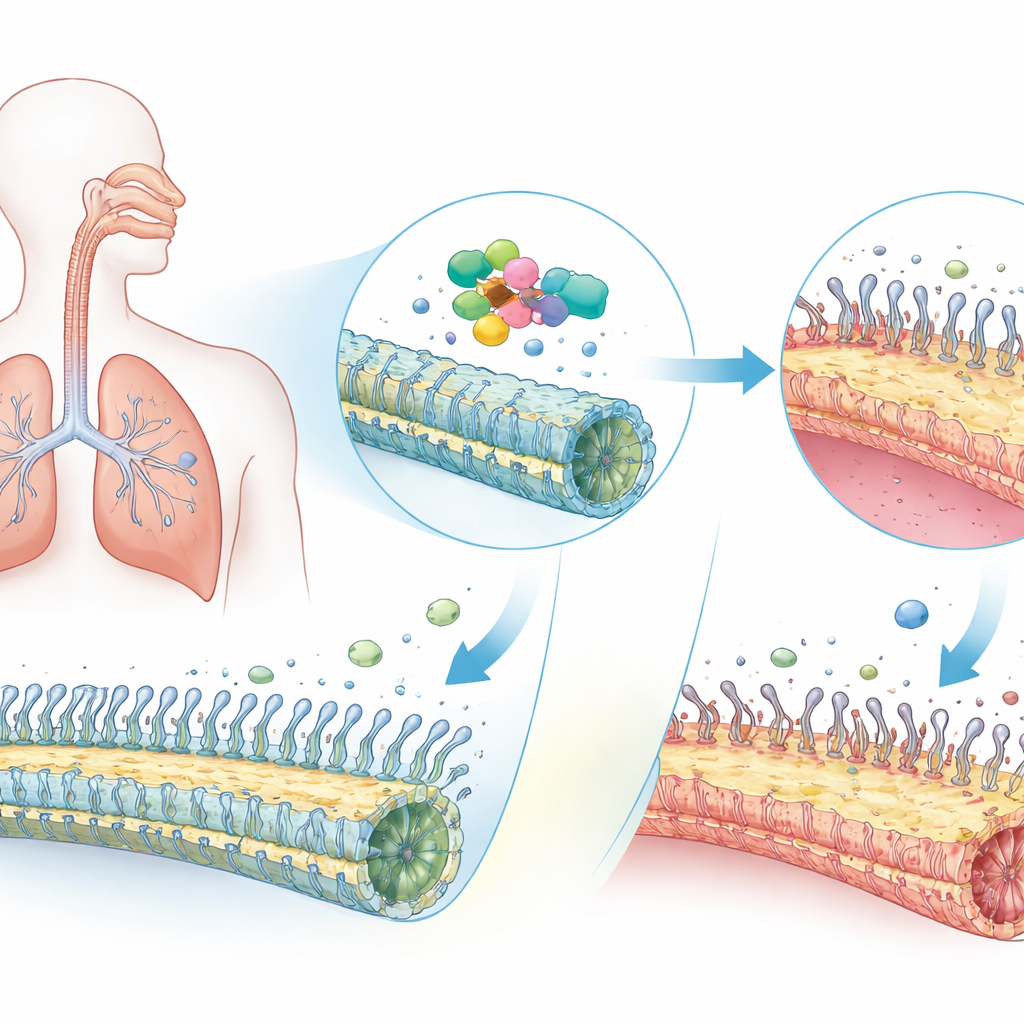
Eine seltene Lungenerkrankung mit großen Folgen
PCD betrifft weltweit etwa eine von einigen tausend Personen und tritt oft bereits im Kindesalter auf. Weil die Zilien in Nase und Lunge nicht effektiv schlagen können, bleiben Schleim und eingeschlossene Mikroben zurück, was chronische Nasennebenhöhlenprobleme, Ohrinfektionen und fortschreitende Lungenschäden begünstigt. Viele Patienten haben außerdem Situs inversus, bei dem Herz und andere Organe spiegelverkehrt angeordnet sind — ein Hinweis darauf, dass Zilien in der Embryonalentwicklung die frühe Körperachse nicht korrekt gesteuert haben. Schon länger ist bekannt, dass Fehler in Dutzenden verschiedener Gene PCD verursachen können. ODAD1 gehört zu diesen Genen und hilft, die molekularen Motoren zu verankern, die die peitschenartigen Bewegungen jeder Zilie antreiben. Wie sich ODAD1‑Defekte jedoch im Gewebe der menschlichen Atemwege auswirken, war bislang nicht vollständig geklärt.
Ein fehlerhaftes Gen in Patienten‑Zellen verfolgen
Die Forscher untersuchten neun Personen aus sieben Han‑chinesischen Familien, die die klassischen Anzeichen von PCD zeigten: Atemprobleme im Neugeborenenalter, lebenslanges produktives Husten, häufige Lungeninfektionen und ungewöhnlich niedrige Stickstoffmonoxidwerte in ausgeatmeter Luft, ein klinisches Kennzeichen der Erkrankung. Genetische Analysen identifizierten vier schädliche Varianten des ODAD1‑Gens, darunter eine zuvor unbekannte. Alle fehlerhaften Varianten führten in Nasenepithelzellen zu einer starken Reduktion oder zum Fehlen des normalen ODAD1‑Proteins. Bei Hochgeschwindigkeitsaufnahmen der Zilien aus Patientenproben ersetzten schwaches, unkoordiniertes Flimmern die sonst glatte, wellenförmige Schlagbewegung. In aus diesen Zellen gezüchteten Labor‑Kulturen, die eine dünne, zilienbedeckte Atemwegsoberfläche an einer Luft‑Flüssig‑Grenze nachbilden, zeigte sich dieselbe träge, unkoordinierte Bewegung.
In den gebrochenen Mikromaschinen
Um zu sehen, was physisch fehlte, setzten die Wissenschaftler leistungsfähige bildgebende Verfahren ein. Konventionelle Elektronenmikroskopie und moderne Kryo‑Elektronenmikroskopie zeigten, dass die äußeren Motorbausteine (ODAs) und deren Andockstellen in den Zilien der Patienten vollständig fehlten. In einigen Fällen waren auch andere wichtige Strukturen im Kern der Zilie fehlplatziert oder fehlgebildet. Diese Defekte erklären, warum die Zilien nicht genug Kraft erzeugen, um Schleim zu bewegen. Doch der Schaden beschränkte sich nicht auf die Zilien selbst. Die Atemwegsoberfläche in patientenabgeleiteten Kulturen wies deutlich weniger multizilientragende Zellen auf, und die verbliebenen Zellen waren unregelmäßig verteilt und vergrößert, mit Zilien, die in verschiedene Richtungen zeigten. Überraschenderweise war die Anzahl der internen „Basalkörper“ pro Zelle — die Keime, aus denen Zilien wachsen — normal, was nahelegt, dass das Problem in der Organisation dieser Zellen an der Gewebeoberfläche lag und nicht in der Anzahl der gebildeten Zilien.
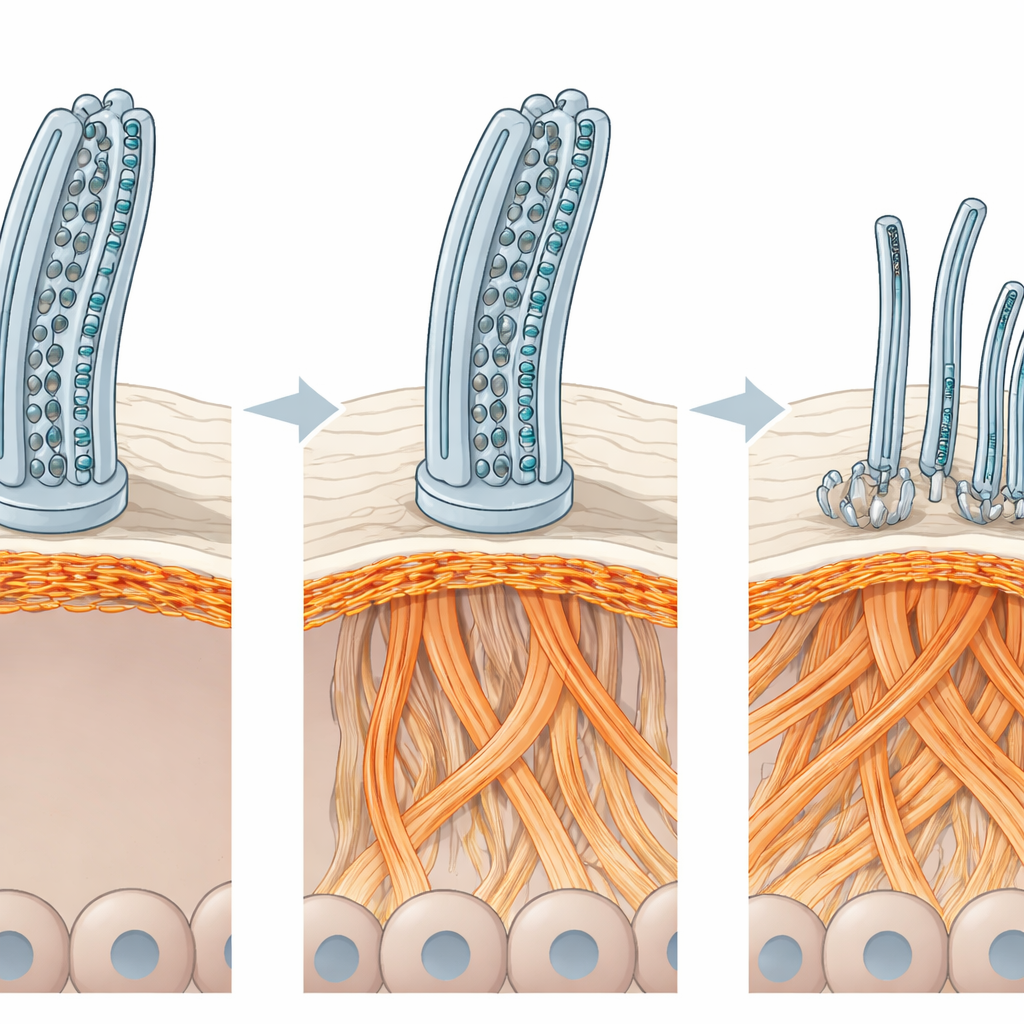
Das innere Gerüst der Zelle wird umverdrahtet
Um herauszufinden, was diese Organisation störte, quantifizierte das Team Tausende von Proteinen in den patientenabgeleiteten Atemwegskulturen. Während viele zilienspezifische Proteine vermindert waren, waren mehrere Proteine, die mit Aktin verbunden sind — den vielseitigen Fasern, die einen Großteil des zellulären Gerüsts bilden — vermehrt vorhanden. Die Bildgebung der Aktinfilamente bestätigte eine dramatische Umgestaltung dieses Gerüsts: verdickte Aktinbündel an der Spitze multizilientragender Zellen, komprimierte Netzwerke an Zellgrenzen und verklumpte Bereiche tiefer im Gewebe. Diese Veränderungen waren nicht auf die genetischen Hintergründe der Patienten beschränkt; das künstliche Ausschalten von ODAD1 in gesunden Zellen führte zur gleichen Aktinumverdrahtung und zum Verlust multizilientragender Zellen. Wenn die Forschenden den Aktinaufbau mit einem kleinen Wirkstoff sanft störten, erholten sich Anzahl und Oberflächeinteilung der multizilientragenden Zellen teilweise, und die Zilien wurden zahlreicher und besser ausgerichtet — blieben jedoch ohne die Andockfunktion von ODAD1 nicht vollständig in ihrer normalen Schlagaktivität wiederherstellbar.
Das fehlende Teil wiederherstellen und Ausblick
Schließlich prüften die Investigatoren, ob das Ersetzen von ODAD1 die Zilienbewegung wiederbeleben könnte. Sie züchteten „apikal‑außen“ Atemwegs‑Organoide — kleine, hohle Kugeln aus Atemweggewebe mit nach außen gerichteten Zilien — aus Patienten‑Zellen und nutzten einen lentiviralen Vektor, um eine funktionsfähige ODAD1‑Kopie einzuschleusen. Das eingeführte Protein siedelte sich korrekt in den Zilien an, stellte die fehlenden Motor‑Andockstellen wieder her und brachte die Zilienschlagbewegung nahezu auf normale Geschwindigkeit und Muster zurück. Zusammengenommen zeigen diese Ergebnisse, dass der Verlust von ODAD1 die Atemwege auf zweifache Weise schädigt: Er deaktiviert direkt das Motorensystem der Zilien und verwirrt indirekt das Aktin‑Gerüst, das die zilientragende Oberfläche formt. Für Patienten deutet diese doppelte Erkenntnis auf eine zweigleisige therapeutische Perspektive hin — Gentherapien zur Behebung des primären Motorendefekts und sicherere Aktin‑modulierende Strategien, um einen gesunden Belag aus Zilien wiederaufzubauen, der die Lungen erneut freihalten kann.
Zitation: Huo, C., Luo, T., Yang, S. et al. Loss-of-function variants in ODAD1 disrupt ODA docking and induce actin cytoskeletal remodeling in primary ciliary dyskinesia. Cell Discov 12, 25 (2026). https://doi.org/10.1038/s41421-026-00875-8
Schlüsselwörter: primäre ziliäre Dyskinesie, ODAD1, bewegliche Zilien, Aktin-Zytoskelett, Gentherapie