Clear Sky Science · de
Integrierte räumliche Profilierungs-Pipeline zur Bestimmung von TME-Architekturen in archivierten klinischen Proben mit der CmTSA-Superplex-Technologie
Warum die verborgene Landschaft um Tumoren wichtig ist
Krebs wächst nicht isoliert. Er ist umgeben von einer lebhaften Nachbarschaft aus Immunzellen, Stützzellen, Blutgefäßen und narbenähnlichem Gewebe, die zusammen die „Mikroumgebung“ des Tumors bilden. Dieser Artikel stellt eine praktikable Methode vor, um diese verborgene Landschaft detailreich mit standardmäßigen Gewebeproben aus Krankenhäusern zu kartieren. Indem gezeigt wird, welche Zelltypen nebeneinander vorkommen und wie sie sich zu hilfreichen oder schädlichen Nachbarschaften organisieren, könnte die Methode Ärzten helfen, besser vorherzusagen, wie sich der Krebs eines Patienten verhalten wird und welche Behandlungen am erfolgversprechendsten sind.
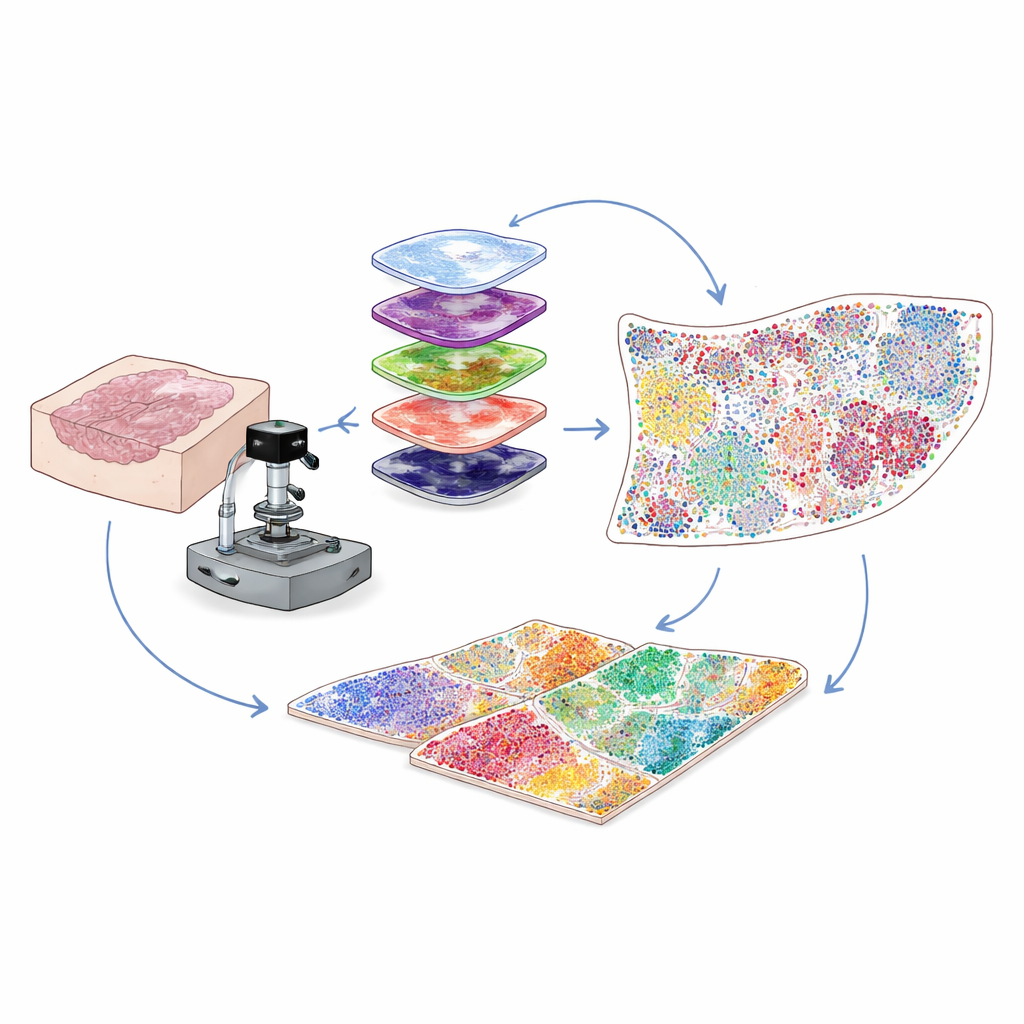
Mehr sehen in alltäglichen Krankenhausproben
Die meisten klinischen Krebsproben werden als dünne Schnitte aus paraffineingebettetem Gewebe konserviert, bekannt als FFPE-Blöcke, die jahrelang gelagert werden können. Diese Proben sind ein Schatz für die Forschung, doch ein technisches Problem hielt Wissenschaftler zurück: Solche Proben zeigen eine natürliche Hintergrundfluoreszenz, die die schwachen Signale vieler wichtiger Proteine überdeckt. Die Autorinnen und Autoren lösten dieses Problem, indem sie intensive, sorgfältig abgestimmte Beleuchtung mit einer milden chemischen Behandlung kombinierten, um die Hintergrundfluoreszenz selektiv zu löschen, ohne Gewebe oder Proteinziele zu schädigen. Dieser hybride optisch-chemische „Bleaching“-Schritt verbessert die Bildklarheit deutlich und erlaubt es, schwache Proteinsignale zu detektieren, die sonst verloren gingen.
Dutzende Proteintags auf derselben Scheibe darstellen
Um zu verstehen, welche Zellen vorhanden sind und was sie tun, färben Forschende Gewebe mit Antikörpern, die an spezifische Proteine binden. Traditionelle Multiplex-Verfahren haben entweder Probleme mit schwachen Signalen für seltene Proteine oder können nur eine begrenzte Anzahl von Markern gleichzeitig verfolgen. Hier verwendet das Team einen Ansatz namens zyklische Tyramid-Signalverstärkung (cyclic tyramide signal amplification). In jeder Runde wird eine kleine Gruppe von Markern eingefärbt und enzymatisch zu hellen, dauerhaft angehefteten Fluoreszenzpunkten „entwickelt“. Die Antikörper werden dann sanft entfernt, während das Signal erhalten bleibt, der Hintergrund wird erneut gebleicht und die nächste Markerrunde wird hinzugefügt. Durch Wiederholung dieses Zyklus viele Male und Ausrichten der Bilder mithilfe des konstanten Signals der Zellkerne können sie zuverlässig 30 bis 60 verschiedene Proteine auf einer einzelnen Gewebescheibe über ein gesamtes Objektträgerfeld in Einzelzellauflösung visualisieren.
Bunte Bilder in ein Zell-für-Zell-Atlas verwandeln
High-Plex-Bilder enthalten Millionen von Pixeln, weit mehr, als ein Mensch mit bloßem Auge analysieren könnte. Die Autorinnen und Autoren bauen daher eine Computer-Vision-Pipeline, die zunächst mit Deep-Learning-Werkzeugen, die ursprünglich für allgemeine Zellsegmentierung entwickelt wurden, jeden Zellkern findet und umreißt. Dann wird je nachdem, wo die Fluoreszenz eines Proteins erscheint — an der Membran, im Zytoplasma oder im Kern — und anhand von logischen Regeln jede Zelle einem Typ oder Subtyp zugeordnet, etwa Tumorzelle, Helfer-T-Zelle, Killer-T-Zelle, B-Zelle, Fibroblast oder andere. Das Ergebnis ist eine digitale Tabelle, die für jede Zelle auf dem Objektträger ihre Identität und ihre exakten Koordinaten auflistet. So wird ein komplexes Bild in eine quantitative Karte verwandelt, die zeigt, wer sich wo in der Tumormikroumgebung befindet.
Aufdeckung zellulärer Nachbarschaften, die das Ergebnis prägen
Zellen agieren selten allein; entscheidend ist, welche Nachbarn sie haben. Um das zu erfassen, testen die Forschenden verschiedene Wege, lokale Nachbarschaften um jede Zelle zu definieren, und entscheiden sich für einen radiusbasierten Netzwerkansatz. Stellen Sie sich vor, um jede Zelle wird ein kleiner Kreis gezogen — etwa so dick wie ein menschliches Haar — und man listet auf, wer darin lebt. Indem Zellen gruppiert werden, deren umliegende Kreise ähnliche Mischungen von Nachbarn aufweisen, identifiziert die Methode wiederkehrende „funktionale Nischen“, wie immunreiche Zonen, fibroblastendominierte Barrieren oder tumordominierte Regionen. Die Anwendung dieser Strategie auf Kolongewebe zeigt, dass radiusbasierte Nachbarschaften besser mit bekannten anatomischen Strukturen übereinstimmen als alternative Methoden. In Zervixkarzinomproben von Patientinnen mit guten gegenüber schlechten Verläufen findet das Team, dass immunzellreiche Nischen bei gutem Outcome nahe dem Tumorrand gruppiert sind, während Patientinnen mit schlechterem Verlauf dicke, fibroblastendichte Zonen aufweisen, die Tumorzellen umschließen und offenbar angreifende Immunzellen abschirmen.
Von räumlichen Karten zu maßgeschneiderten Therapien
Durch die Kombination hochwertiger, kostengünstiger Färbung vieler Proteine mit robuster Bildanalyse liefert diese Arbeit eine durchgängige Pipeline, die auf große Mengen standardmäßiger Krankenhausproben angewendet werden kann. Die Methode verwandelt konserviertes Gewebe in detaillierte Karten darüber, wie Tumor-, Immun- und Stromazellen sich anordnen und interagieren. Für Laien lautet die Kernbotschaft: Nicht nur die Zelltypen, sondern ihre präzisen Nachbarschaftsmuster sind entscheidend dafür, wie sich ein Krebs verhält. Diese Plattform könnte Forschenden helfen, schützende Immun-Hotspots zu identifizieren, supprimierende Zellbarrieren zu erkennen und letztlich genauere Prognosen sowie feinere, auf Immuntherapie abgestimmte Behandlungsstrategien zu unterstützen.
Zitation: Xiao, C., Zhou, R., Chen, Q. et al. Integrative spatial profiling pipeline for determining TME architectures in archival clinical specimens using CmTSA superplex technology. Cell Discov 12, 16 (2026). https://doi.org/10.1038/s41421-026-00874-9
Schlüsselwörter: Tumormikroumgebung, räumliche Proteomik, multiplexe Bildgebung, Krebsimmunologie, Einzelzellanalyse