Clear Sky Science · de
ATGL macht hepatozelluläre Karzinomzellen gegenüber genotoxischen Medikamenten empfindlicher, indem es den Acetylierungs-/Phosphorylierungsstatus von p53 moduliert
Den Fettabbau zur Schwachstelle des Krebses machen
Die Standardchemotherapie bei Leberkrebs versagt oft, weil Tumorzellen bemerkenswert gut darin sind, DNA‑Schäden zu überstehen. Diese Studie untersucht einen überraschenden Verbündeten innerhalb genau dieser Krebszellen: ein Enzym, das gespeicherte Fette abbaut. Durch Erhöhen dieses Enzyms, genannt ATGL, stellten die Forschenden fest, dass Lebertumorzellen weniger DNA‑Reparatur betreiben und stattdessen zur Selbstzerstörung gedrängt werden. Die Arbeit offenbart eine bislang verborgene Verbindung zwischen dem Lipidstoffwechsel der Krebszellen und ihrer Reaktion auf starke DNA‑schädigende Medikamente und deutet auf neue Möglichkeiten hin, bestehende Therapien effektiver zu machen.
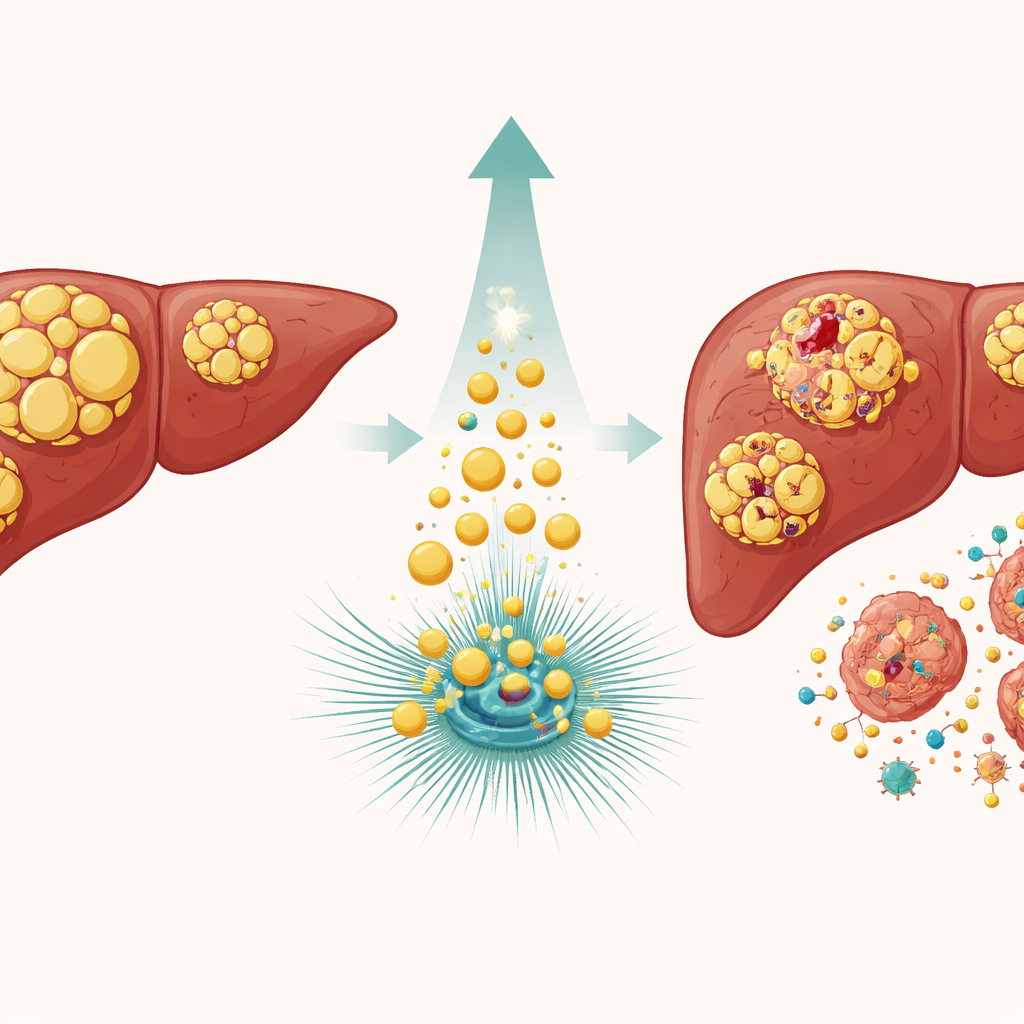
Warum Lebertumoren harten Medikamenten widerstehen
Leberkrebs, insbesondere das hepatozelluläre Karzinom, zählt weltweit zu den häufigsten und tödlichsten Tumorarten. Viele Patientinnen und Patienten erhalten Medikamente, die DNA schädigen, wie Etoposid und Doxorubicin, in der Hoffnung, Krebszellen in eine tödliche Krise zu stürzen. Dennoch entkommen diese Zellen häufig, indem sie ihr Wachstum pausieren und Reparatursysteme aktivieren, die von einem Wächterprotein namens p53 gesteuert werden. Wird der Schaden behoben, teilen sich die Zellen weiter; ist das nicht möglich, kann p53 auch den programmierten Zelltod auslösen. Das zentrale Rätsel ist, was p53 zugunsten von Reparatur oder Selbstzerstörung entscheidet und warum manche Tumoren so hartnäckig therapieresistent bleiben.
Ein fettspaltendes Enzym verschiebt die Waage
Das Team konzentrierte sich auf ATGL, ein Enzym, das gespeicherte Fette in winzigen zellulären Behältern, den Lipidtröpfchen, abbaut. In Lebertumoren sind die ATGL‑Spiegel in der Regel niedriger als im gesunden Gewebe, und frühere Arbeiten deuteten darauf hin, dass eine erzwungene ATGL‑Expression das Tumorwachstum bremst. In der vorliegenden Studie veränderten die Forschenden Leberkrebszelllinien so, dass sie entweder mehr ATGL produzierten oder dessen Menge verringerten, und setzten sie dann DNA‑schädigenden Medikamenten aus. Zellen mit erhöhtem ATGL zeigten deutlich mehr Anzeichen gebrochener DNA, während Zellen mit reduziertem ATGL weniger Schäden aufwiesen. Das Hemmen der ATGL‑Spaltaktivität mit einem spezifischen Inhibitor oder die Expression einer funktionsunfähigen Mutante beseitigte diese gesteigerte Empfindlichkeit — ein Beleg dafür, dass die fettspaltende Aktivität des Enzyms selbst entscheidend ist.
Die Entscheidung der Zelle umlenken: Reparatur oder Tod
Auf tieferer Ebene untersuchten die Wissenschaftler p53, das nach DNA‑Schäden wie ein molekularer Verkehrspolizist wirkt. Das Verhalten von p53 wird durch kleine chemische Markierungen an spezifischen Stellen gesteuert. In ATGL‑reichen Zellen führten genotoxische Medikamente dazu, dass p53 mehr von einer Markierung (Acetylgruppen) und vergleichsweise weniger von einer anderen (Phosphatgruppen) trägt. Diese Verschiebung begünstigte die Aktivierung von Genen, die den Zelltod fördern, etwa Puma, und dämpfte Gene wie p21 und GADD45, die normalerweise den Zellzyklus anhalten und die DNA‑Reparatur unterstützen. Infolgedessen konnten ATGL‑reiche Zellen selbst nach Entfernung des Medikaments die Marker für DNA‑Schäden nicht beseitigen und gingen in die Apoptose statt in die Erholung über.
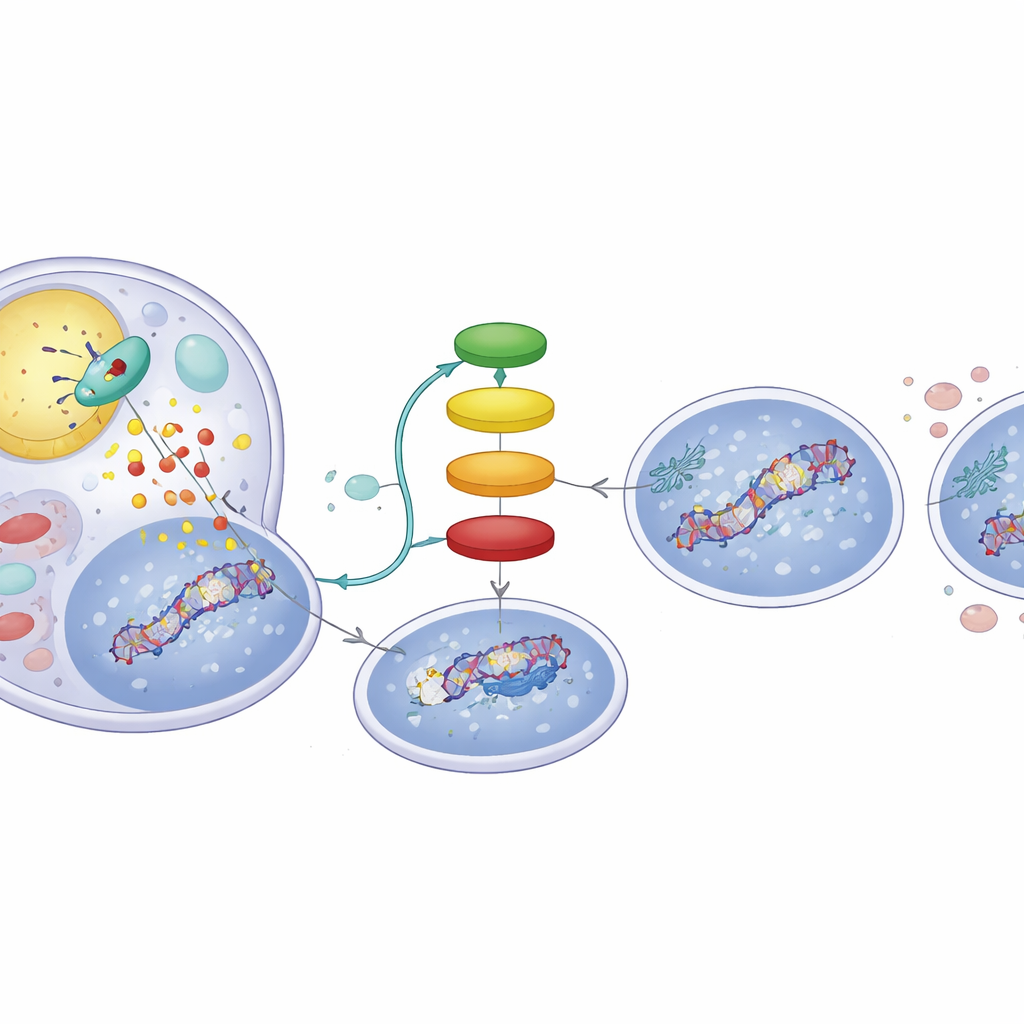
Eine fetttreibende Signalkette innerhalb der Tumorzellen
Wie verändert der Fettabbau die Markierungen von p53? Die Abbauprodukte von ATGL sind freie Fettsäuren, die als Botenstoffe wirken können. Die Studie zeigt, dass diese Fettsäuren einen nukleären Rezeptor namens PPARα aktivieren, der wiederum die Aktivität von p300 steigert — einem Protein, das Acetylgruppen auf p53 anbringt. Als die Forschenden eine PPARα‑aktivierende Verbindung verwendeten, stellten sie das ATGL‑hohe Verhalten nach: verstärkte DNA‑Schadenssignale und ein p53‑Profil, das zur Apoptose neigt. Umgekehrt beseitigte die Blockade von p300 die ATGL‑induzierten Veränderungen an p53 und reduzierte die DNA‑Schäden, was unterstreicht, dass eine ATGL → PPARα → p300‑Kette zentral für diesen Schalter ist. Analysen von Hunderten menschlicher Lebertumoren aus öffentlichen Datensätzen bestätigten diesen Zusammenhang: Tumoren mit höherer ATGL‑Expression zeigten ebenfalls stärkere PPARα‑ und p300‑Signaturen sowie eine erhöhte Expression p53‑gesteuerter Gene.
Welche Bedeutung das für zukünftige Therapien haben könnte
Kurz gesagt zeigt die Studie: Wenn Leberkrebszellen dazu angeregt werden, gespeicherte Fette über ATGL zu verbrennen, sind sie weniger geneigt, durch Chemotherapie induzierte DNA‑Schäden zu reparieren, und neigen eher zu geordnetem Zelltod. Das eröffnet zwei praktische Möglichkeiten: Die Messung von ATGL‑Spiegeln könnte helfen vorherzusagen, welche Patientinnen und Patienten besser auf genotoxische Medikamente ansprechen, und das Fördern von ATGL‑Aktivität oder seines nachgeschalteten PPARα‑Wegs könnte zusammen mit bestehenden Chemotherapien genutzt werden, um Resistenz zu überwinden. Zwar sind weitere Tests an Tieren und in klinischen Studien nötig, doch die Arbeit vermittelt eine eindrückliche Botschaft: Bei Leberkrebs könnte das „Innen‑Verschlanken“ der Tumorzellen sie auf mikroskopischer Ebene verwundbarer gegenüber lebensrettenden Medikamenten machen.
Zitation: Castelli, S., De Cristofaro, A., Desideri, E. et al. ATGL sensitizes hepatocellular carcinoma cells to genotoxic drugs by modulating p53 acetylation/phosphorylation status. Cell Death Discov. 12, 164 (2026). https://doi.org/10.1038/s41420-026-03048-4
Schlüsselwörter: hepatozelluläres Karzinom, ATGL, DNA-Schadensantwort, p53-Signalweg, Lipidstoffwechsel bei Krebs