Clear Sky Science · de
DNA-Polymerase kappa, durch Ptbp2 stabilisiert, interagiert mit MRE11 und fördert genomische Instabilität bei Leukämie
Wie Leukämiezellen gebrochene DNA behalten und trotzdem überleben
Unsere DNA ist ständig Angriffen ausgesetzt, doch gesunde Zellen sind normalerweise sehr gut darin, Schäden zu erkennen und zu reparieren. Bei Leukämie lernen jedoch einige Zellen, mit gebrochener, instabiler DNA zu leben — und verwandeln diese Instabilität sogar in einen Überlebensvorteil. Diese Studie deckt ein molekulares „Teamwork“ zwischen einem Spleißprotein (Ptbp2), einer speziellen DNA-Kopiervorstufe (DNA-Polymerase kappa, oder Polk) und einem Schadenssensor (MRE11) auf, das Leukämiezellen dabei hilft, gerade genug Schäden zu beheben, um zu überleben, während sie die genetische Unordnung anhäufen, die das Fortschreiten des Krebses antreibt.
Ein versteckter Helfer in Leukämiezellen
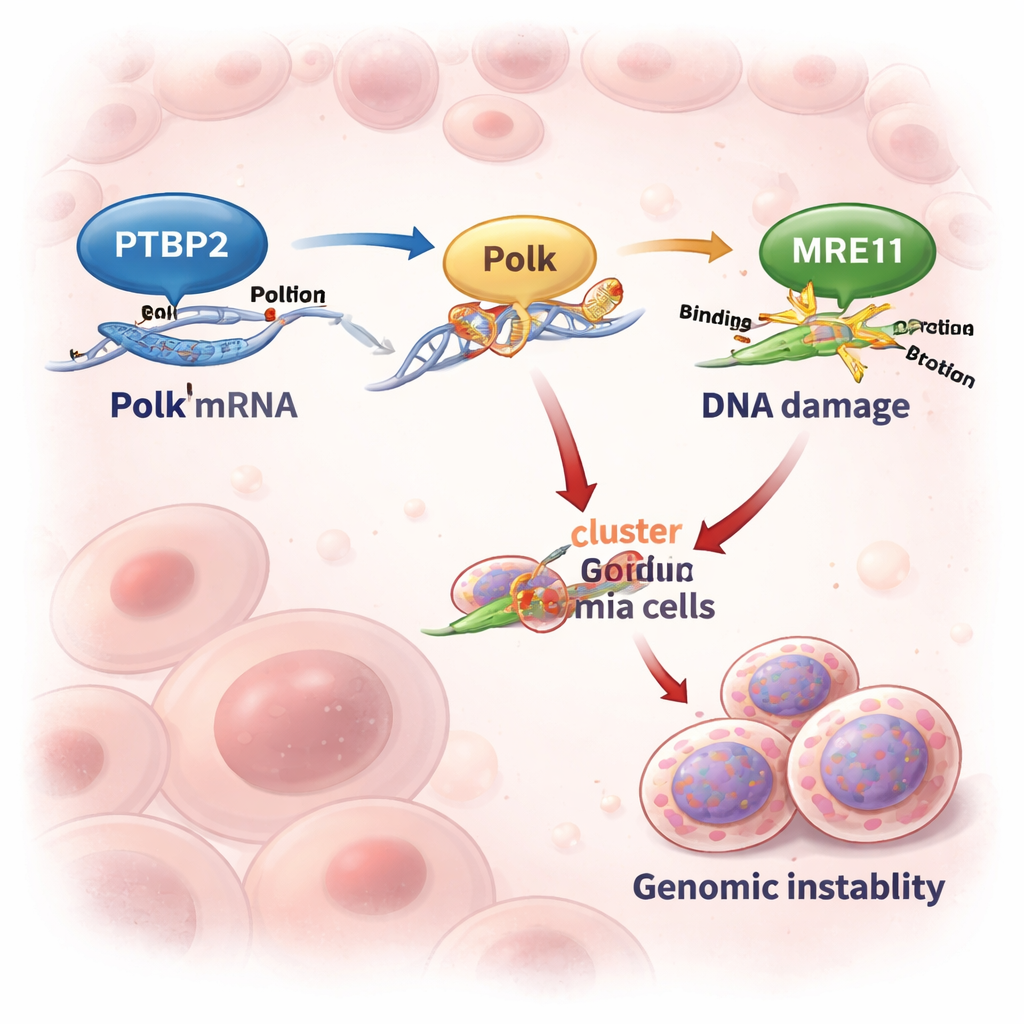
Die Forschenden konzentrierten sich auf die chronische myeloische Leukämie (CML), eine Blutkrebserkrankung, die meist durch das Fusionsgen BCR::ABL1 getrieben wird. Während moderne Medikamente, die BCR::ABL1 blockieren, in frühen Stadien gut wirken, sprechen viele Patientinnen und Patienten in der aggressiven „Blastenkrebs“-Phase schlecht darauf an. Frühere Arbeiten zeigten, dass Ptbp2, ein Protein, das an RNA bindet und die Nachrichtenverarbeitung beeinflusst, durch BCR::ABL1 hochreguliert wird und in der CML onkogen wirkt. Hier entdeckte das Team, dass Ptbp2 sich an das 3′-UTR-Ende der Polk-mRNA heftet und sie vor Abbau schützt. Infolgedessen produzieren Leukämiezellen mehr Polk-Protein, wenn Ptbp2 erhöht ist.
Aktivierung einer fehleranfälligen DNA-Kopiermaschine
Polk ist eine „Backup“-DNA-Polymerase, die über beschädigte DNA kopieren kann, wenn die üblichen Replikationsmaschinen ins Stocken geraten. Diese Fähigkeit kann gestresste Zellen retten — sie hat aber ihren Preis, denn Polk ist fehleranfällig und kann Mutationen einführen. In Zelllinien und Patientenproben aus fortgeschrittener CML stiegen und fielen Ptbp2- und Polk-Spiegel gemeinsam. Wenn Forschende Ptbp2 in Leukämiezellen ausschalteten, fielen die Polk-Spiegel stark ab und die Polk-RNA zerfiel nahezu doppelt so schnell. Die Wiedereinführung von Polk in Ptbp2-defiziente Zellen stellte ihr Verhalten wieder her und zeigte, dass Ptbp2 hier vor allem Polk stabilisiert und so dessen Präsenz und Aktivität sicherstellt.
Reparatur von Schäden — aber nicht perfekt
Um zu sehen, wie dieses Duo die DNA-Reparatur beeinflusst, behandelten die Forschenden Zellen mit Hydroxyurea, einem Medikament, das die DNA-Replikation blockiert und häufig bei CML-Patienten eingesetzt wird. Zellen ohne Ptbp2 erlitten deutlich mehr DNA-Schäden, sichtbar als lange „Kometenschweife" und helle γH2AX-Foci — Kennzeichen gebrochener Chromosomen. Diese geschädigten Zellen starben häufiger. Im Gegensatz dazu tolerierten Zellen mit hohem Ptbp2- und Polk-Spiegel das Medikament besser, reparierten Schäden effizienter und überlebten, obwohl ihre Reparatur schlampig war. Die Überexpression von Polk in Ptbp2-Knockout-Zellen hob diese Empfindlichkeit auf und bestätigte, dass die Ptbp2–Polk-Partnerschaft Leukämiezellen hilft, Replikationsstress zu überstehen und Apoptose zu vermeiden.
Ein DNA-Schadensnetzwerk, das Instabilität begünstigt
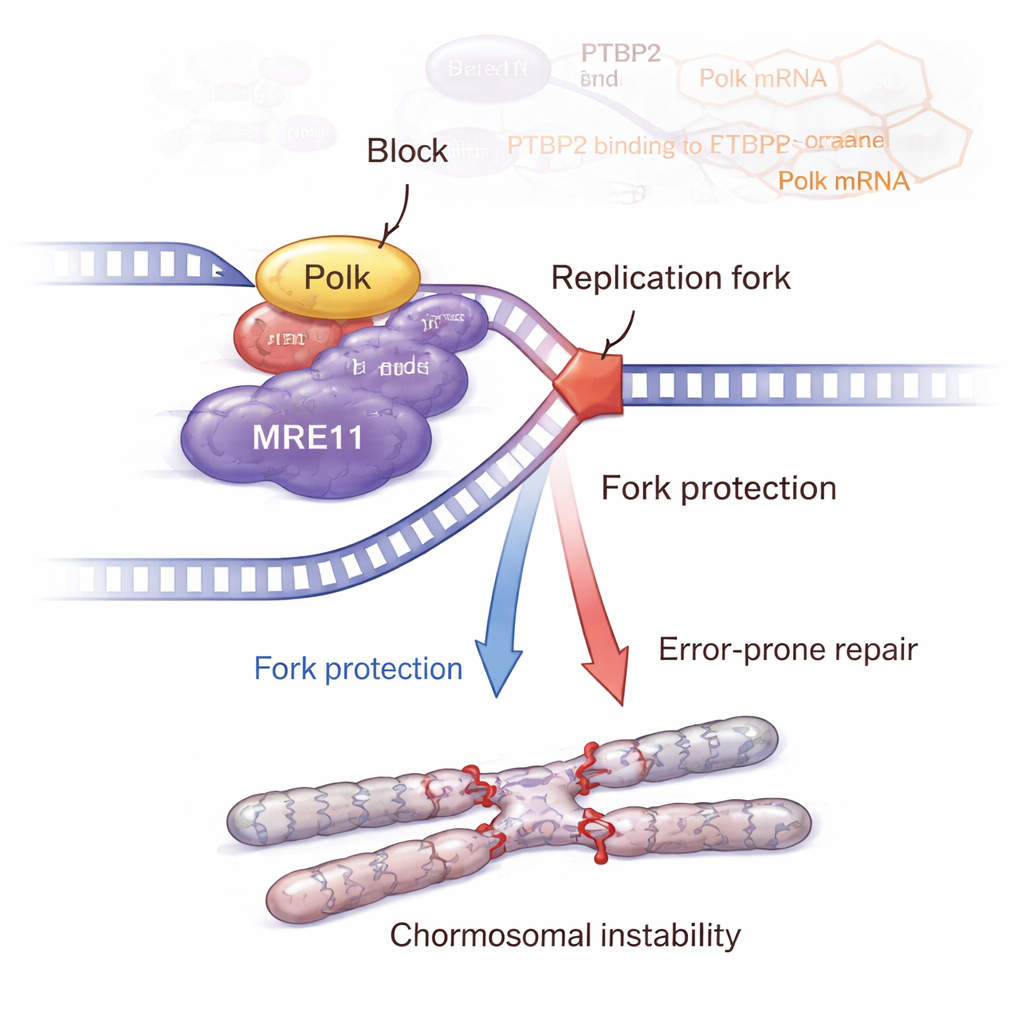
Die Geschichte endet nicht bei Polk. Das Team zeigte, dass Polk physisch mit MRE11 interagiert, einem Schlüsselmitglied des MRN-Komplexes, der DNA-Brüche erkennt und die ATM–CHK2-Schadensantwort aktiviert. Wenn Ptbp2 entfernt wurde, sanken Polk, MRE11-Spiegel und dessen Aktivität, und die ATM–CHK2-Signalgebung schwächte sich ab. Die Wiedereinsetzung von Polk stellte MRE11 und dessen Aktivierung wieder her. Detaillierte DNA-Faser-Experimente zeigten, dass Ptbp2 und Polk gestoppte Replikationsgabeln weitgehend über MRE11 vor Rückumsätzen schützen. Die Hemmung von MRE11 mit einem Wirkstoff untergrub diesen Gabelschutz und verstärkte DNA-Schäden. Paradoxerweise sammelten Zellen mit aktivem Ptbp2–Polk–MRE11-Signalweg mehr chromosomale Auffälligkeiten an, wie Schwesterchromatidaustausche, Brüche, Spalten, multipolare Spindeln und mehrkernige Riesen-Zellen — klassische Zeichen genomischer Instabilität, die aggressiveren Krebs fördern können.
Von Mäusen zu möglichen neuen Therapien
In Mausmodellen erzeugten Leukämiezellen mit intaktem Ptbp2 größere, abnormalere Tumoren als Zellen ohne Ptbp2. Gewebe dieser Mäuse zeigten höhere Werte von Ptbp2, Polk, des Proliferationsmarkers Ki-67 und verzerrte Zellteilungsstrukturen. In einem separaten, von BCR::ABL1 getriebenen CML-ähnlichen Mausmodell verstärkte zusätzliches Ptbp2 Polk und erhöhte die Zahl atypischer teilender Zellen sowie invasiver Leukämieherde in Milz und Leber, was auf ein schnelleres Fortschreiten der Erkrankung hindeutet. Zusammengenommen deuten diese Befunde darauf hin, dass die Achse Ptbp2–Polk–MRE11–ATM–CHK2 es Leukämiezellen erlaubt, intensiven DNA-Stress zu überstehen und gleichzeitig schädliche Mutationen anzusammeln.
Warum das für Patientinnen und Patienten wichtig ist
Für Laien lautet die Kernaussage: Einige Leukämiezellen entziehen sich der Kontrolle, indem sie auf einem Drahtseil balancieren: Sie reparieren ihre DNA gerade so weit, dass sie überleben, aber nicht so vollständig, dass Mutationen verhindert werden. Ptbp2 stabilisiert Polk, das sich mit MRE11 verbündet, um gestresste DNA zu schützen und Schadenssignale aufrechtzuerhalten — doch diese Reparatur ist unvollkommen und fördert genetisches Chaos. Da fortgeschrittene CML und andere Krebsarten offenbar von diesem fragilen Gleichgewicht abhängen, könnte das Anvisieren von Ptbp2 oder seiner Kontrolle über Polk Zellen aus dem Überlebensmodus in den Selbstzerstörungsmodus kippen und einen vielversprechenden neuen Therapieansatz bieten, besonders in der schwer behandelbaren Blastenkrebs-Phase.
Zitation: Lama, S., Barik, B., IS, S. et al. DNA polymerase kappa stabilized by Ptbp2 interacts with MRE11 and promotes genomic instability in leukemia. Cell Death Discov. 12, 96 (2026). https://doi.org/10.1038/s41420-026-02951-0
Schlüsselwörter: chronische myeloische Leukämie, genomische Instabilität, DNA-Reparatur, DNA-Polymerase kappa, PTBP2