Clear Sky Science · de
Hemmung von RRM1 macht Adenokarzinom der Lunge empfindlicher für Decitabin-Behandlung
Aus einem lauwarmen Medikament einen stärkeren Verbündeten machen
Lungenkrebs bleibt eine der tödlichsten Krebsarten, und viele Patientinnen und Patienten laufen früher oder später aus effektiven Behandlungsoptionen heraus. Ärztinnen und Ärzte hoffen schon lange, dass Medikamente, die die DNA von Krebszellen behutsam umprogrammieren anstatt lediglich teilende Zellen zu vergiften, hilfreich sein könnten. Ein solches Medikament, Decitabin, wirkt gut bei Blutkrebserkrankungen, hat sich aber bei soliden Tumoren wie Lungenkrebs als enttäuschend erwiesen. Diese Studie stellt eine einfache, praktische Frage mit großen Folgen: Gibt es einen Weg, Lungentumoren dazu zu bringen, endlich auf Decitabin anzusprechen — mithilfe von Werkzeugen, die wir bereits verstehen?
Warum ein bewährtes Medikament in soliden Tumoren versagt
Decitabin ist ein täuschend ähnlicher Baustein zu einem der DNA-Bausteine. Wenn es sich beim Kopieren in die DNA einer Zelle einschleicht, kann es abnorme chemische Markierungen entfernen, die schützende Gene stilllegen, darunter Tumorsuppressoren und Immungen-Gene. Bei Leukämien hilft das, Zellen in einen gesünderen Zustand zurückzusetzen. In Lungentumoren hingegen wirkt das Medikament kaum. Die Autorinnen und Autoren vermuteten, dass das Problem nicht in der Wirkungsweise von Decitabin liegt, sondern darin, dass nur sehr wenig davon tatsächlich in die DNA solider Tumorzellen eingebaut wird. Durch Messung winziger Mengen des in die DNA eingebauten Medikaments in vielen Krebszelllinien bestätigten sie, dass Zellen, die mehr Decitabin integrierten, deutlich empfindlicher gegen das Medikament waren.
Ein zellulärer Türsteher, der das Medikament blockiert
Um herauszufinden, was den Eintritt des Medikaments in die DNA begrenzt, untersuchten die Forschenden Gene, die an der Verarbeitung von Nukleosiden beteiligt sind — den Rohstoffen für DNA. Ein Enzym mit dem Namen RRM1 fiel besonders auf. RRM1 ist Teil eines Mechanismus, der gewöhnliche Bausteine in die aktiven Formen umwandelt, die beim DNA-Bau verwendet werden. Beim Lungenadenokarzinom war dieses Enzym in den Tumoren im Vergleich zum normalen Lungengewebe ungewöhnlich stark ausgeprägt, und Patientinnen und Patienten mit niedrigeren RRM1-Spiegeln hatten tendenziell eine längere Überlebenszeit. In einem Panel von Krebszelllinien ging ein höherer RRM1-Spiegel mit einer geringeren Decitabin-Einlagerung einher, was stark darauf hindeutet, dass dieses Enzym als Türsteher wirkt und das Medikament verdrängt.
Den Türsteher entwaffnen, um dem Medikament zu helfen
Das Team fragte dann, was passiert, wenn sie RRM1 teilweise ausschalten. Mit genetischen Werkzeugen reduzierten sie RRM1 in Lungenkrebszellen, ohne die Zellen sofort abzutöten. Allein hatte diese Reduktion nur einen milden Einfluss auf das Wachstum. In Kombination mit niedrigen Dosen Decitabin war die Wirkung jedoch dramatisch: Kolonien von Lungenkrebszellen schrumpften in Kultur deutlich, und Tumoren wuchsen in Mäusen deutlich langsamer. Wichtig ist, dass diese wirksamen Dosen gut verträglich waren, ohne erkennbare Schädigungen von Blut, Leber oder Nierenfunktion bei den Tieren. Auf molekularer Ebene erlaubte die Blockade von RRM1, dass mehr Decitabin in die DNA eingebaut wurde, was zu einem stärkeren Verlust des methylierenden Enzyms DNMT1 und einem größeren Abfall der globalen DNA-Methylierung führte. Dies wiederum weckte Tumorsuppressor-Gene, die zuvor ausgeschaltet gewesen waren.
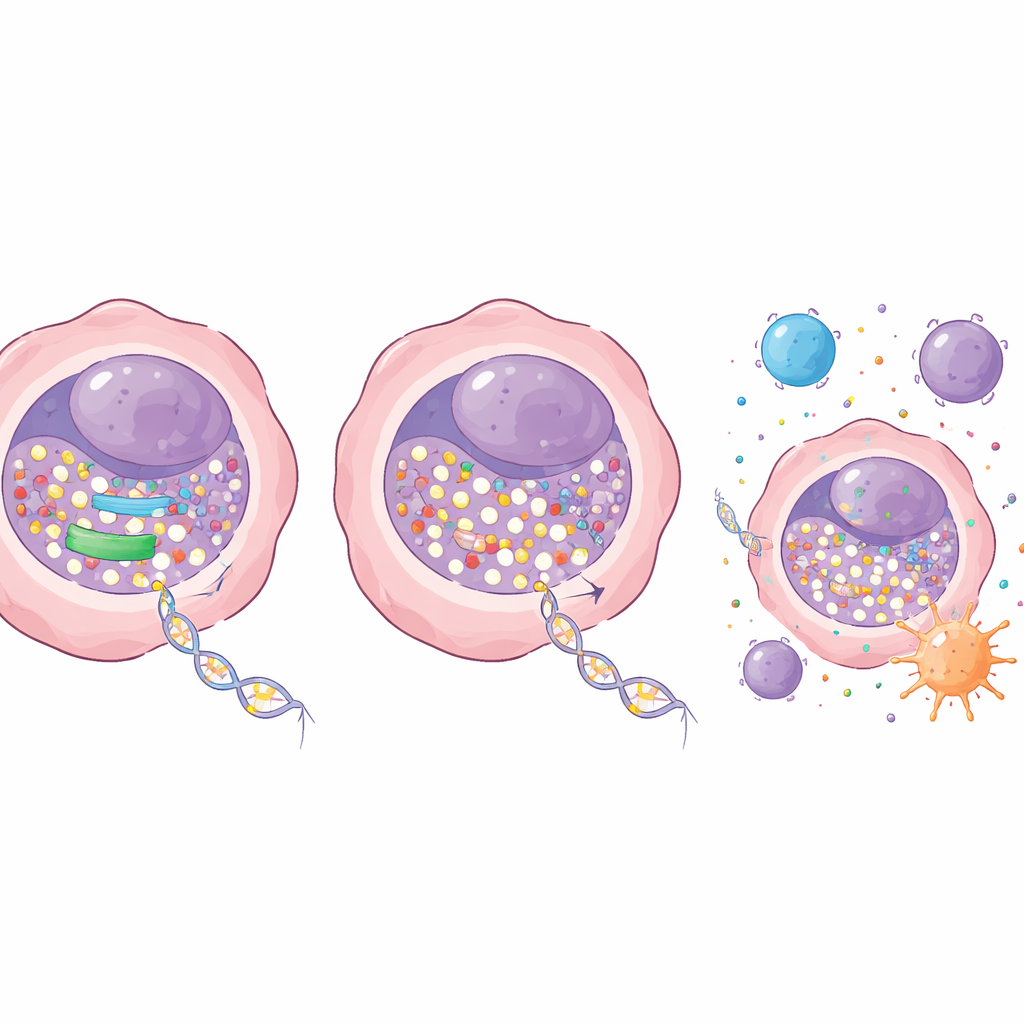
Das Immun-Alarmzentrum innerhalb der Tumoren einschalten
Über die Verlangsamung der Zellteilung hinaus veränderte die Kombinationstherapie, wie Krebszellen mit dem Immunsystem interagieren. Mehr Decitabin in der DNA verstärkte Signale für DNA-Schäden in den Zellen und trieb sie in Richtung programmierter Zelltod. Gleichzeitig erhöhte es die Aktivität eines inneren Alarmsystems rund um den STING-Weg, der fehlplatziertes DNA erkennt und antiviralen-ähnliche Immunantworten auslöst. Wenn RRM1 blockiert war, aktivierte Decitabin diesen Weg und seine nachgeschalteten Gene stärker, einschließlich solcher, die Immunzellen anlocken und stimulieren. In Mausmodellen des Lungenkrebses mit intaktem Immunsystem erzeugte die Kombination von Decitabin mit einem RRM1-Inhibitor eine stärkere Tumorkontrolle als eine der beiden Therapien allein, ohne zusätzliche offensichtliche Toxizität. Die Autorinnen und Autoren fanden außerdem, dass diese Enzym-Blockierungsstrategie Decitabin spezifisch verstärkt und bei einem verwandten Wirkstoff, Azacitidin, sogar entgegenwirken kann — ein Hinweis darauf, dass die Wahl der richtigen Partner entscheidend ist.
Was das für Patientinnen und Patienten bedeuten könnte
In der Summe zeichnet die Arbeit ein klares Bild: Ein überaktives DNA-Baustein-Enzym in Lungentumoren begrenzt, wie viel Decitabin sein Ziel erreicht. Durch partielle Hemmung dieses Enzyms werden Krebszellen gezwungen, mehr des Medikaments anstelle ihrer üblichen Bausteine zu verwenden. Dieser Wandel ermöglicht es, dass niedrige Decitabin-Dosen schützende Gene effektiver wieder aktivieren, die DNA von Krebszellen schädigen und Immunabwehrmechanismen wecken — und das bei guter Verträglichkeit in Tiermodellen. Für Patientinnen und Patienten deutet dies einen realistischen Weg nach vorn an: Die Umwidmung oder Verfeinerung von RRM1-Inhibitoren in Kombination mit niedrig dosiertem Decitabin und möglicherweise modernen Immuntherapien, um ein einst unzureichendes Medikament zu einem nützlichen Bestandteil der Lungenkrebsbehandlung zu machen.
Zitation: Jiang, N., Liu, J., Vaghasia, A. et al. RRM1 inhibition sensitizes lung adenocarcinoma to decitabine treatment. Cell Death Dis 17, 275 (2026). https://doi.org/10.1038/s41419-026-08522-6
Schlüsselwörter: Lungenadenokarzinom, Decitabin, DNA-Methylierung, Ribonukleotidreduktase, Krebsimmuntherapie