Clear Sky Science · de
Semaphorin 6D treibt antitumorale Typ-I-Interferon-Antworten an und programmiert das Tumormikromilieu beim kolorektalen Krebs um
Warum diese Forschung für Menschen mit Darmkrebs wichtig ist
Kolorektales Karzinom gehört weltweit zu den häufigsten Krebstodesursachen, unter anderem weil viele Tumoren gegenüber den heutigen Therapien resistent sind, einschließlich moderner Immuntherapien. Diese Studie enthüllt ein natürliches „Bremssystem“ in Darmtumoren, das oft abgeschaltet ist, und zeigt, wie dessen Reaktivierung das Immunsystem wieder in den Kampf holen kann. Das Verständnis dieses versteckten Schalters könnte Ärzten helfen, Prognosen besser zu stellen und Kombinationstherapien zu entwickeln, die Immuntherapien für deutlich mehr Patienten wirksam machen.
Ein stiller Wächter in Tumorzellen
Im Mittelpunkt der Arbeit steht ein Molekül namens Semaphorin 6D (SEMA6D), das ursprünglich für die Führung des Nervenwachstums und die Prägung des sich entwickelnden Herzens bekannt war. Die Forschenden fanden heraus, dass SEMA6D beim kolorektalen Krebs wie ein Tumorsuppressor wirkt: In gesundem Darmgewebe ist es vorhanden, in Tumorgewebe sind die Werte dagegen deutlich reduziert. In mehreren Patientendatensätzen und Tumorproben war niedriger SEMA6D-Spiegel mit größeren Tumoren, tieferer Invasion, mehr Metastasen und deutlich schlechterem Überleben verknüpft. Dieses Muster blieb bestehen, selbst wenn andere klinische Faktoren berücksichtigt wurden, was darauf hinweist, dass SEMA6D ein unabhängiger Marker für die Aggressivität eines kolorektalen Tumors ist.
Wie Tumoren diesen Schutz ausschalten
Das Team fragte dann, warum SEMA6D in Tumoren so häufig fehlt. Sie entdeckten, dass das Gen häufig durch eine chemische Modifikation namens Promoter-Hypermethylierung abgeschaltet wird — zusätzliche chemische Markierungen im Kontrollbereich des Gens, die wie molekulares Klebeband über einem Lichtschalter wirken. Durch detaillierte DNA-Kartierung zeigten sie, dass wichtige Abschnitte der SEMA6D-Kontrollregion in Krebszellen stark methyliert sind, in normalen Darmzellen jedoch nicht. Behandelten sie Krebszellen mit einem Demethylierungsmittel, das in hämatologischen Tumoren eingesetzt wird, wurden die Methylmarken entfernt und die SEMA6D-Produktion wiederhergestellt. Die niedrigsten SEMA6D-Werte fanden sich in Subtypen des Darmkrebses, die bereits für starke DNA-Methylierung, hohe genetische Instabilität und ausgeprägte Metastasierungsneigung bekannt sind, was die Verbindung zwischen diesem Stilllegungsmechanismus und aggressiver Erkrankung stärkt.
Vom Wachstumshemmer zum Immunverstärker
Die Wiederherstellung von SEMA6D veränderte das Tumorverhalten auf zwei Ebenen. Erstens, auf der Ebene der Krebszellen: Durch erzwungene Überexpression von SEMA6D verlangsamte sich ihr Wachstum, ihre Wander- und Invasionsfähigkeit nahm ab, und Merkmale des epithelial–mesenchymalen Übergangs, eines Programms, das Tumoren beim Streuen hilft, wurden rückgängig gemacht. In Zellkulturen und dreidimensionalen Organoiden aus Patientenproben bildeten Zellen mit mehr SEMA6D weniger und kleinere Kolonien und zeigten verstärkte Zeichen programmierter Zellteilung. In Mäusen wuchsen Tumoren, die so verändert waren, dass sie mehr SEMA6D produzierten, langsamer und bildeten weniger Lungen- und Lebermetastasen, während das Herunterregulieren von SEMA6D den gegenteiligen Effekt hatte. Zweitens, auf der Ebene des Immunsystems: SEMA6D-reiche Tumoren in immunkompetenten Mäusen enthielten deutlich mehr CD4- und CD8-T-Zellen — die Hauptangriffsgruppen der adaptiven Immunität —, während SEMA6D-arme Tumoren relativ leer von diesen Abwehrzellen waren. Als die Forschenden T-Zellen depletionierten, verschwand der wachstumshemmende Effekt von SEMA6D größtenteils, was zeigt, dass ein großer Teil seiner Wirksamkeit aus der Aktivierung des Immunsystems stammt.
Entschlüsselung des internen Alarmwegs
Tiefergehend kartierte die Studie die molekularen Schritte, die SEMA6D mit der Immunaktivierung verbinden. An der Oberfläche der Tumorzellen signalisiert SEMA6D über einen Partnerrezeptor namens Plexin A4. Im Zellinneren interagiert dieses Duo physisch mit einem Protein namens IRF9, einem zentralen Bestandteil der Maschinerie, die auf Typ-I-Interferone reagiert — dieselben antiviralen Alarmsignale, die Zellen zur Bekämpfung von Infektionen nutzen. Wenn SEMA6D vorhanden ist und Plexin A4 intakt ist, werden IRF9 und seine Partner aktiviert, schalten Reihen von interferon-stimulierten Genen an und helfen der Tumorzelle, Signale auszusenden, die T-Zellen anlocken und aktivieren. Das Entfernen von SEMA6D oder Plexin A4 unterbricht diese Kette und dämpft den Alarm; die Wiederherstellung von IRF9 kann den Effekt teilweise retten. In Mäusen zeigten Tumoren mit aktivem SEMA6D–Plexin A4–IRF9-Signalweg mehr infiltrierende T-Zellen und niedrigere Werte des Proliferationsmarkers Ki-67, was mit einem stärkeren immunologischen Druck auf den Tumor übereinstimmt.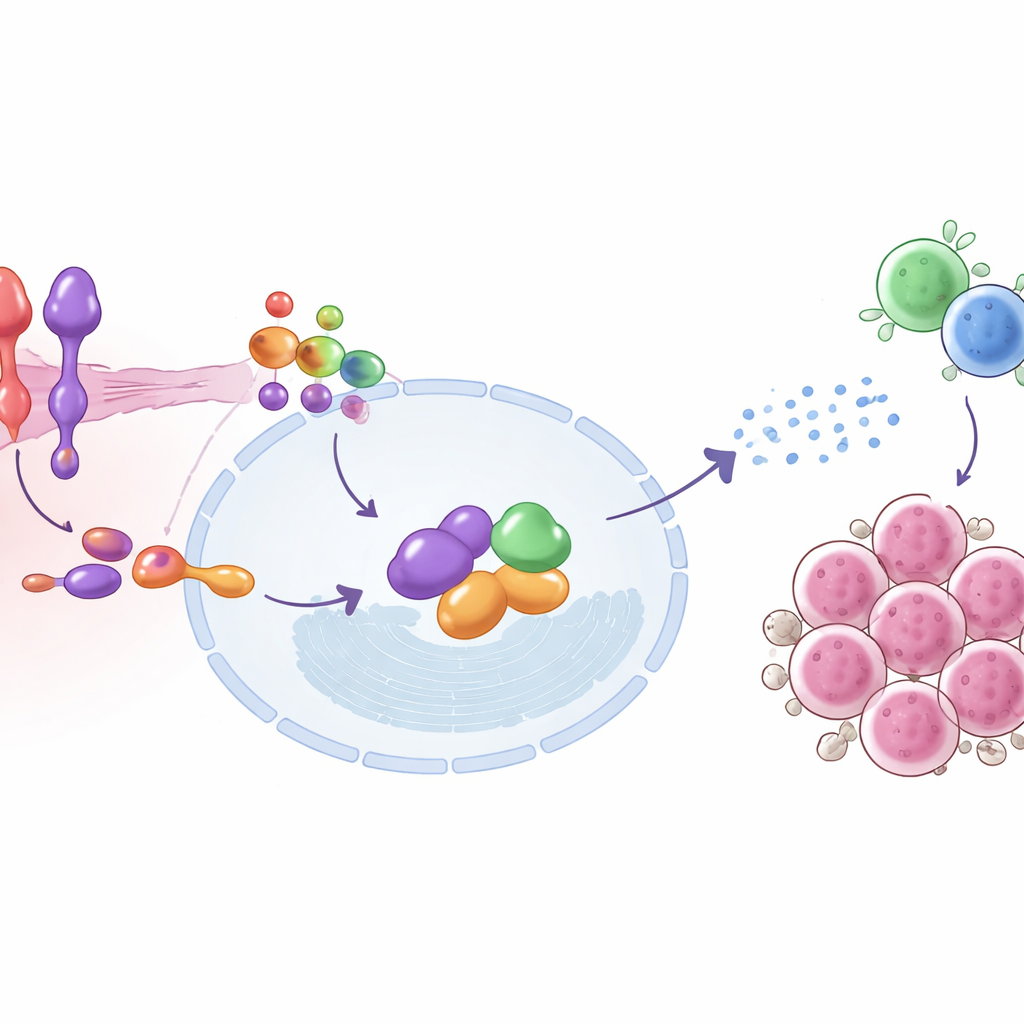
Wiedererwecken der Immunität mit Kombinationstherapie
Da SEMA6D durch Methylierung stummgeschaltet wird, prüften die Autoren, ob ein hypomethylierses Medikament es in lebenden Tumoren reaktivieren und so die Reaktion auf Checkpoint-Blockade verbessern könnte. In Maus-Kolontumoren, die zuerst mit Decitabin und anschließend mit einem Anti–PD-1-Antikörper behandelt wurden, wuchsen die Tumoren deutlich langsamer als bei einer Einzelbehandlung. Die Kombination erhöhte SEMA6D-Spiegel, steigerte die Aktivität des Interferonwegs, verringerte die Zellproliferation und förderte die T-Zell-Infiltration. Diese Ergebnisse legen nahe, dass epigenetische Wirkstoffe, indem sie Methylierungs"schlösser" von immunrelevanten Genen wie SEMA6D entfernen, immunologisch "kalte" Tumoren in "wärmere" verwandeln können, die gegenüber Checkpoint-Inhibitoren anfälliger sind.
Was das für die zukünftige Versorgung bedeutet
Für Laien bedeutet die Schlussfolgerung: Einige Darmkrebserkrankungen verstecken sich vor dem Immunsystem, indem sie ein internes Warnsignal chemisch abschalten. Diese Arbeit identifiziert SEMA6D sowohl als dieses Signal als auch als vielversprechenden therapeutischen Ansatzpunkt. Die Messung von SEMA6D und seines Methylierungsstatus könnte helfen, Tumoren zu klassifizieren, Prognosen zu stellen und Therapieentscheidungen zu unterstützen. Ebenso wichtig ist, dass die Studie eine klare biologische Begründung dafür liefert, DNA-Demethylierungsmittel mit Immuntherapie zu kombinieren, um die immunologische Überwachung bei Patienten wiederherzustellen, deren Tumoren derzeit nicht ansprechen. Klinische Studien sind zwar noch erforderlich, doch könnte diese Strategie eines Tages die Vorteile der Immuntherapie für eine wesentlich größere Gruppe von Patienten mit kolorektalem Krebs öffnen.
Zitation: Shi, W., Zhang, F., Sun, WQ. et al. Semaphorin 6D drives anti-tumor type I interferon responses to reprogram the tumor microenvironment in colorectal cancer. Cell Death Dis 17, 256 (2026). https://doi.org/10.1038/s41419-026-08478-7
Schlüsselwörter: kolorektales Karzinom, Tumormikroumgebung, epigenetische Therapie, Typ-I-Interferon, Tumorimmunologie