Clear Sky Science · de
Loss-of-function-Varianten im HPDL beeinträchtigen die menschliche kortikale Entwicklung durch Veränderungen der Mitochondrienfunktion
Warum winzige Zellkraftwerke für wachsende Gehirne wichtig sind
Die meisten Menschen denken bei der Gehirnentwicklung an Gene und Vernetzung. Diese Studie zeigt, dass ein oft übersehener Faktor — die kleinen Kraftwerke in unseren Zellen, die Mitochondrien — ebenfalls die Gehirnformung beeinflussen kann. Anhand seltener kindlicher Bewegungsstörungen, die mit dem Gen HPDL verknüpft sind, zeigen die Autoren, wie fehlerhafte Energieproduktion die sich entwickelnde Großhirnrinde verkleinern kann, eine Region, die für Bewegung, Denken und Verhalten entscheidend ist.
Eine seltene Bewegungsstörung als Einblick in das Gehirnwachstum
Bei einigen Kindern mit Veränderungen im HPDL-Gen entwickelt sich eine hereditäre spastische Paraplegie — eine Erkrankung, die Steifheit und Schwäche in den Beinen verursacht, häufig verbunden mit Anfällen, Entwicklungsverzögerung und in schweren Fällen einem kleineren als normalen Gehirn (Mikrozephalie). Obwohl bekannt war, dass das HPDL-Protein in Mitochondrien sitzt, war seine genaue Funktion — und warum sein Verlust das Gehirn schädigt — unklar. Die Forscher nutzten mehrere humane Zellmodelle, darunter tumorähnliche Nervenzellen und aus Hautproben der Patienten gezüchtete Gehirnzellen, um zu prüfen, ob HPDL für normale Gehirnentwicklung und mitochondriale Gesundheit erforderlich ist.
Was passiert, wenn HPDL abgeschaltet wird
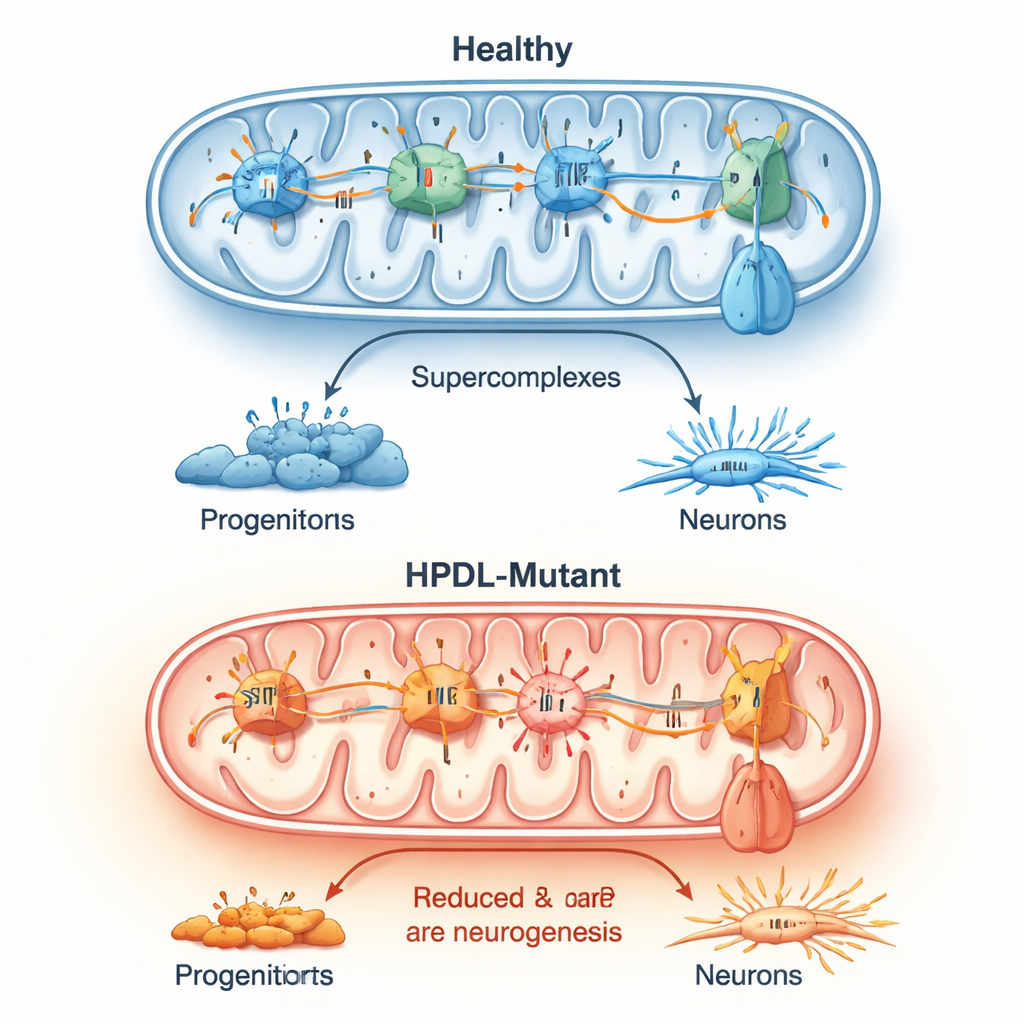
Zuerst schaltete das Team HPDL in einer menschlichen Neuroblastom-Zelllinie mittels CRISPR-Geneditierung aus. Ohne HPDL verloren diese Zellen das Voll-Längen-Protein und zeigten deutliche mitochondriale Probleme. Die großen Verbünde von Atmungs-Ketten-Proteinen, die normalerweise zusammenarbeiten, um Energie zu erzeugen, waren gestört, und Schlüsselkomponenten der Sauerstoffnutzung waren reduziert. Die Zellen verbrauchten weniger Sauerstoff, erzeugten weniger atemwegsgebundene Energie und produzierten mehr reaktive Sauerstoffspezies — schädliche Nebenprodukte, oft als „oxidativer Stress“ bezeichnet. Dennoch nahm die Gesamtzahl der Mitochondrien nicht ab, und die Konzentration von Coenzym Q10, einem wichtigen Molekül für den Energietransfer, war tatsächlich erhöht, was auf einen qualitativen — nicht nur quantitativen — Defekt der Mitochondrienfunktion hindeutet.
Gehirngewebe in der Schale zeigt frühe Überproduktion von Neuronen
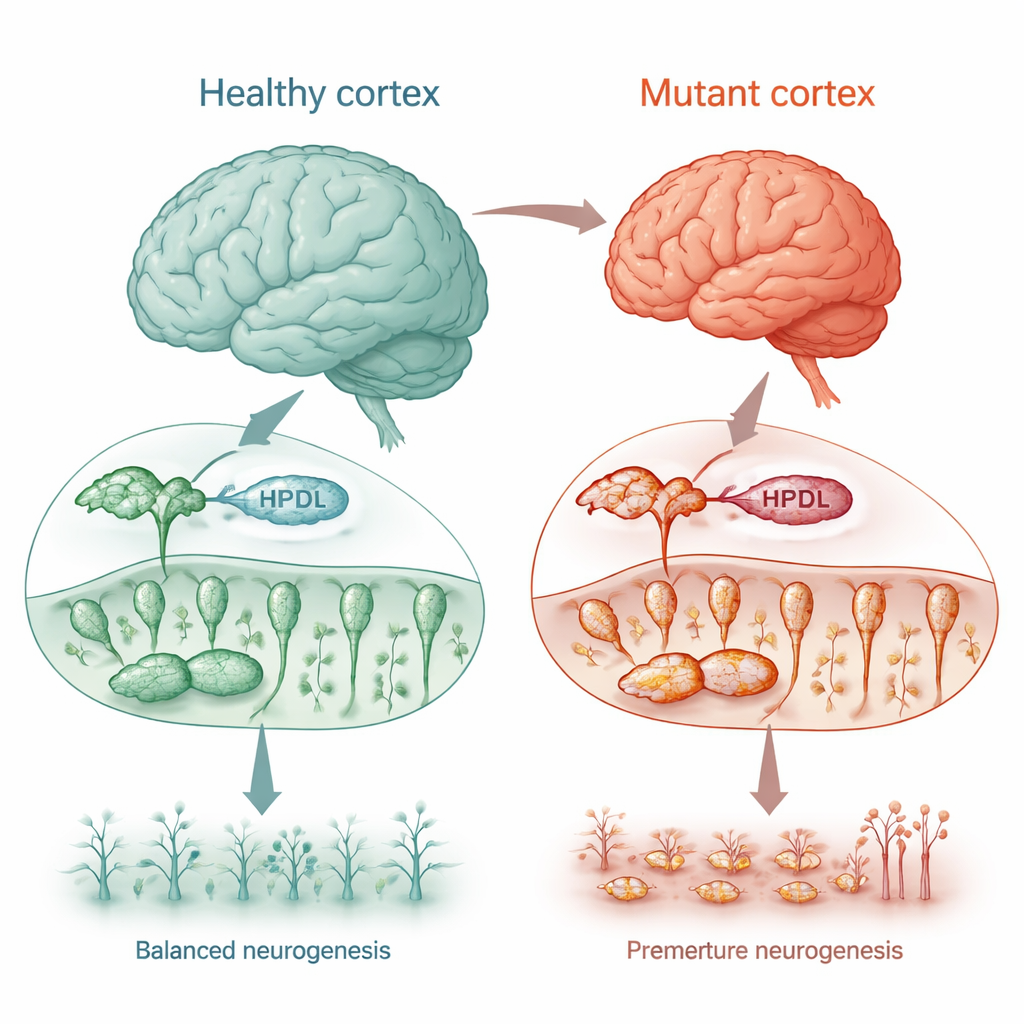
Um zu sehen, wie der Verlust von HPDL die menschliche Gehirnentwicklung beeinflusst, reprogrammierten die Forscher Hautzellen von vier betroffenen Kindern zu induzierten pluripotenten Stammzellen und leiteten sie dann zur Bildung kortikaler Gehirnzellen und dreidimensionaler „Mini-Gehirne“ (Organoide) an. Früh in der Entwicklung, in einer Phase, in der sich die meisten Zellen noch als neuronale Vorläufer teilen sollten, enthielten HPDL-mutante Kulturen bereits mehr reife Neuronen und weniger Vorläuferzellen. Transkriptionsprofile bestätigten dies: Signalwege, die die Neuronbildung antreiben, waren zu früh aktiviert, während Wege, die die Zellteilung aufrechterhalten, heruntergefahren waren. In den Organoiden führte dieser vorzeitige Wechsel von Bausteinen zu fertigen Neuronen zu deutlich kleineren gehirnähnlichen Strukturen, was der Mikrozephalie bei den schwer betroffenen Kindern entspricht.
Beschädigte Kraftwerke und gestresste Zellen
Eine genauere Untersuchung zeigte, dass HPDL-mutante Gehirnzellen eine beeinträchtigte oxidative Phosphorylierung aufwiesen — den Hauptweg, über den Mitochondrien Energie erzeugen. Enzymfärbungen zeigten eine schwächere Aktivität eines wichtigen mitochondrialen Komplexes, während andere Messungen eine veränderte Membranladung über die mitochondriale Membran anzeigten. In vielen mutanten Zellen schien ein entscheidendes Enzym, das normalerweise ATP erzeugt, in umgekehrter Richtung zu arbeiten, um diese Membrandifferenz aufrechtzuerhalten — ein Zeichen tiefgreifender metabolischer Störung. In allen Patientenlinien waren reaktive Sauerstoffspezies konsistent erhöht, und die normalen großen Verbunde der Atmungskettenproteine waren schlechter ausgebildet. Diese mitochondrialen Veränderungen korrelierten eng mit dem Zeitpunkt und dem Ausmaß der vorzeitigen Neuronbildung.
Tests zur Minderung des Stresses
Da oxidativer Stress und gestörte Coenzym-Q10-Chemie zentral erschienen, prüfte das Team, ob Behandlungen, die diese Probleme ansprechen, das Voranschreiten der Neuronbildung verlangsamen könnten. Sie setzten frühe kortikale Kulturen zwei Antioxidantien und 4‑Hydroxybenzoat aus, einem kleinen Molekül, das mit der Coenzym‑Q10‑Synthese verwandt ist. In mehreren patientenabgeleiteten Linien verringerten diese Verbindungen die vorzeitige Neurogenese teilweise, doch die Wirkung hing von der konkreten HPDL‑Mutation ab. Einige Linien sprachen überwiegend auf Antioxidantien an, andere auf den Coenzym‑Q10‑Vorläufer, und eine Linie zeigte gar keine Reaktion. Dieses mutationsspezifische Muster deutet darauf hin, dass personalisierte Behandlungsstrategien für HPDL‑bedingte Störungen nötig sein könnten.
Was das für Kinder und künftige Therapien bedeutet
Vereinfacht gesagt zeigt diese Studie, dass HPDL als Wächter der zerebralen Bausteine während der frühen Entwicklung wirkt. Versagt HPDL, werden Mitochondrien ineffizient und übermäßig gestresst, wodurch Vorläuferzellen zu früh zu Neuronen differenzieren. Der Pool teilender Zellen wird erschöpft, die Großhirnrinde kann nicht ihre volle Größe erreichen, und Verschaltungs‑muster verändern sich, was zu Bewegungsstörungen und weiteren Symptomen beiträgt. Die partielle Rettung durch Antioxidantien und Coenzym‑Q10‑verwandte Verbindungen legt nahe, dass die Anpassung des zellulären Energiegleichgewichts und des oxidativen Stresses eines Tages Kindern mit HPDL‑Mutationen — und möglicherweise auch anderen mitochondriellen Hirnerkrankungen — helfen könnte.
Zitation: Baggiani, M., Desbats, M.A., Naef, V. et al. Loss of function variants in HPDL impair human cortical development via alterations of mitochondrial function. Cell Death Dis 17, 237 (2026). https://doi.org/10.1038/s41419-026-08476-9
Schlüsselwörter: HPDL, Mitochondrien, kortikale Entwicklung, Mikrozephalie, oxidativer Stress