Clear Sky Science · de
MLKL in Leberparenchymzellen fördert Leberkrebs bei muriner, stoffwechselbedingter steatotischer Lebererkrankung
Warum eine Fettleber zu Krebs werden kann
Mit dem Anstieg von Adipositas und Typ‑2‑Diabetes entwickelt eine wachsende Zahl von Menschen auch eine fettleberartige, entzündliche Erkrankung, die heute als metabolisch dysfunktionsassoziierte Steatohepatitis (MASH) bezeichnet wird. Einige dieser Patientinnen und Patienten entwickeln im Verlauf Leberkrebs, doch die Gründe dafür sind noch nicht vollständig geklärt. Diese Studie untersucht ein Protein namens MLKL in Leberzellen und stellt eine einfache, aber weitreichende Frage: Trägt MLKL dazu bei, dass eine kranke, fettleibige Leber in Krebs übergeht, und könnte das Ausschalten dieses Proteins das Risiko für Leberkrebs senken?
Ein genauerer Blick auf Leberschäden in einer modernen Epidemie
Leberkrebs gehört heute weltweit zu den führenden krebsbedingten Todesursachen, und ein großer Teil dieses Anstiegs ist auf Fettlebererkrankungen zurückzuführen, die durch schlechte Ernährung und Stoffwechselstörungen begünstigt werden. Bei MASH sind Leberzellen mit Fett überladen, stehen unter Stress und sind von chronischer Entzündung umgeben. Über Jahre kann dieses Milieu DNA schädigen und es mutierten Zellen ermöglichen, zu Tumoren heranzuwachsen. Das Protein MLKL ist vor allem dafür bekannt, eine gewaltsame Form des Zelltods auszulösen, bei der Zellen aufplatzen und Entzündungen verstärken. Deshalb vermuteten Forscherinnen und Forscher, dass MLKL einer der Schalter sein könnte, der langanhaltende Leberschädigung in einen ausgewachsenen Leberkrebs verwandelt.
Prüfung der Rolle von MLKL in einem Mausmodell für fettleberbedingten Leberkrebs
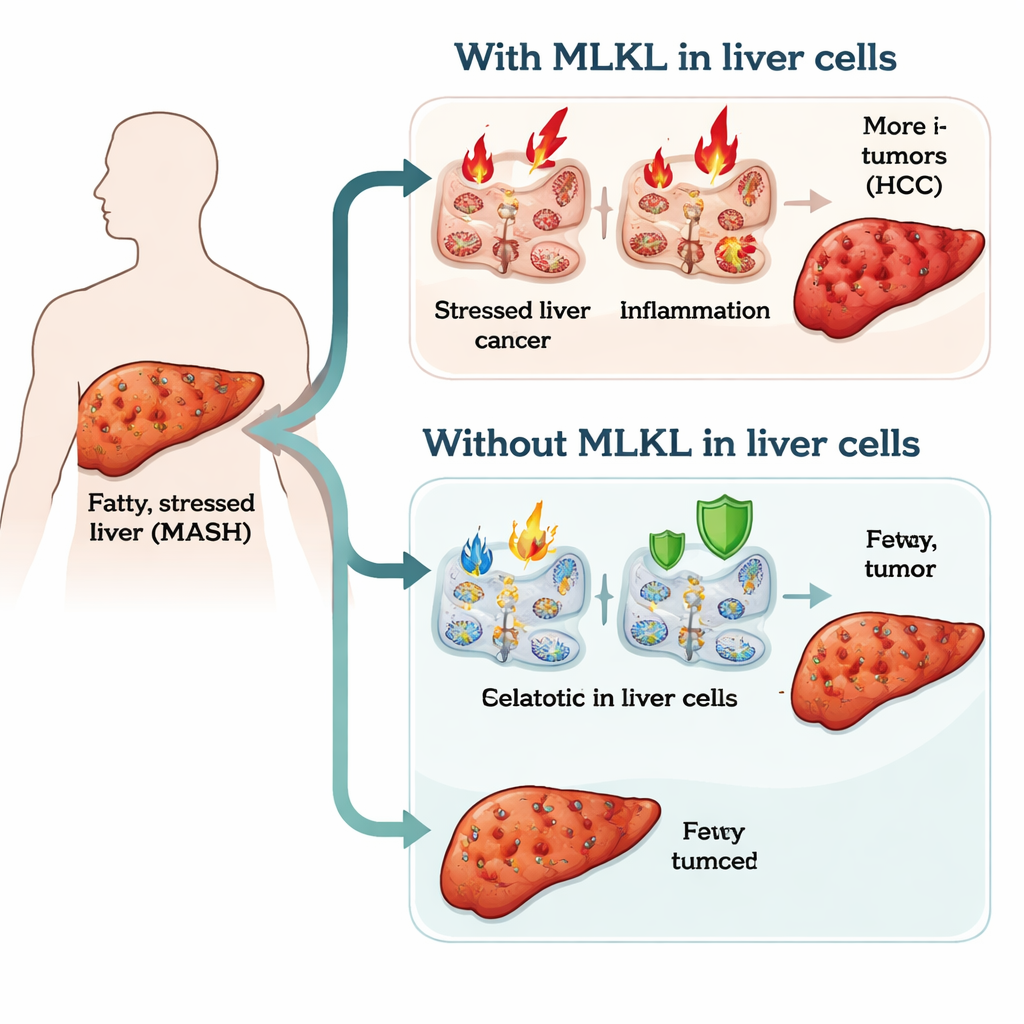
Um die Rolle von MLKL zu untersuchen, erzeugten die Forschenden Mäuse, bei denen MLKL spezifisch aus Leberparenchymzellen entfernt wurde – hauptsächlich den arbeitenden Leberzellen, den Hepatozyten –, während andere Gewebe unverändert blieben. Alle Mäuse wurden kurz nach der Geburt diabetisch gemacht und anschließend entweder mit einer normalen Diät oder einer fettreichen, zuckerreichen Diät gefüttert, die die Ernährungsumgebung nachahmt, die beim Menschen zu MASH führt. Innerhalb von 12 Wochen verursachte diese Diät zuverlässig eine Fettleber, geringe Vernarbung und – wichtig – Lebertumoren, die dem menschlichen hepatozellulären Karzinom ähneln. So konnte das Team normale Mäuse direkt mit Mäusen vergleichen, deren Leberzellen MLKL‑defizient waren, und beobachten, wie sich die Krankheit in jedem Fall entwickelte.
Gleiche Fettleber, aber verzögerte Entzündung und weniger Tumoren
Überraschenderweise verhinderte das Entfernen von MLKL aus Leberzellen nicht die frühen Krankheitsstadien: Beide Mausgruppen nahmen ähnlich zu, lagerten Fett in der Leber ein und entwickelten vergleichbare, milde Fibrose. Die auffälligen Unterschiede zeigten sich in der Geschwindigkeit, mit der Entzündung und Krebs auftraten. Mäuse mit normalem MLKL zeigten früher erhöhte entzündliche Signale in der Leber, mehr aktivierte Immunzellen in Leber und Milz und vergrößerte Milzen – Hinweise auf eine starke, systemische Entzündungsreaktion. Im Gegensatz dazu hatten Mäuse ohne MLKL in Leberzellen eine verzögerte Entzündungsphase und weniger stark aktivierte Immunzellen in frühen Stadien. Im Laufe der Zeit führte dies zu weniger und kleineren Lebertumoren, einer geringeren Rate fortgeschrittener präkanzeröser Veränderungen und einer deutlich reduzierten Inzidenz von vollständigem hepatozellulärem Karzinom.
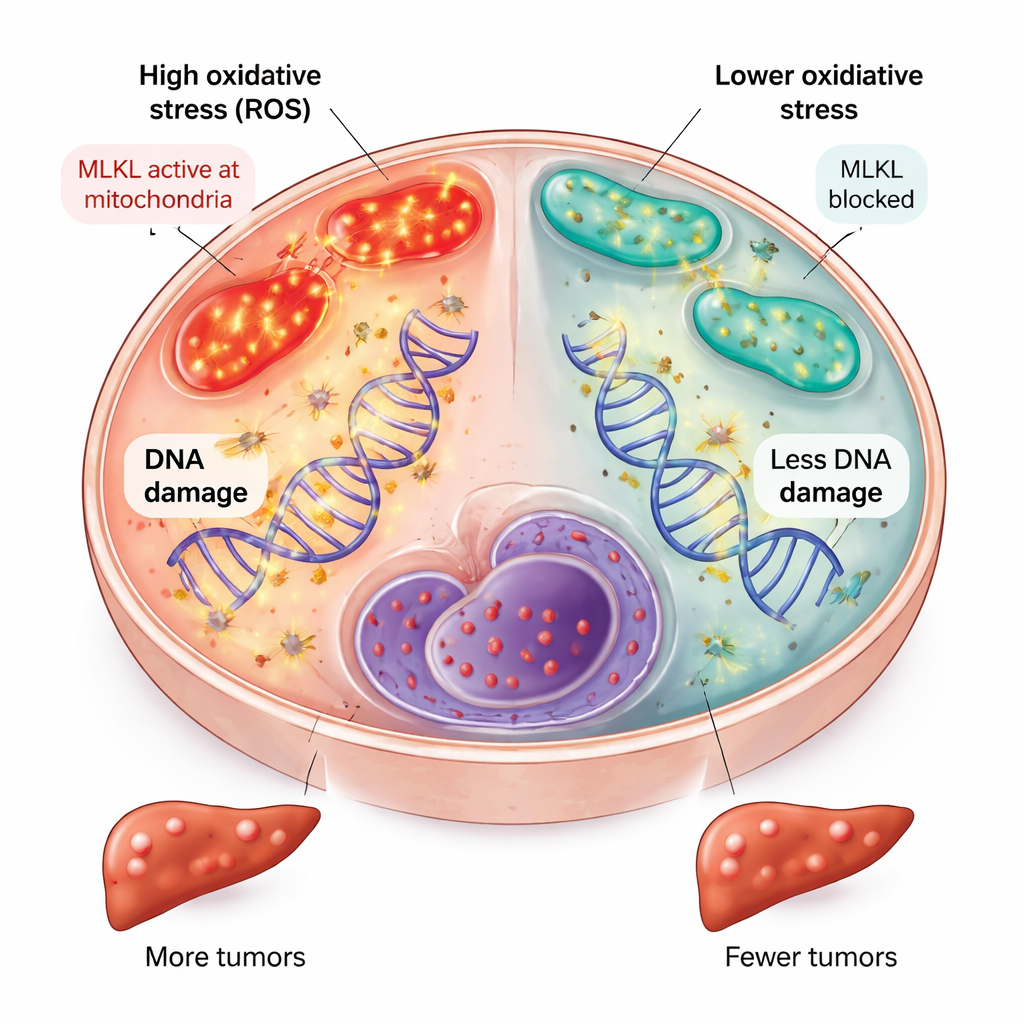
Weniger oxidativer Stress und DNA‑Schäden ohne MLKL
Das Team fragte dann, warum das Fehlen von MLKL in Leberzellen das Fortschreiten zum Krebs verlangsamte. Sie fanden heraus, dass Lebern ohne MLKL früh im Krankheitsverlauf eine geringere Expression von Genen zeigten, die durch oxidativen Stress aktiviert werden, weniger biochemische Anzeichen metabolischen Stresses in bestimmten Lipiden namens Sphingolipide aufwiesen und einen ausgewogeneren Zustand von Coenzym Q in den Mitochondrien, den Energiefabriken der Zelle, hatten. Diese Veränderungen deuten alle auf weniger schädliche reaktive Sauerstoffspezies hin. Konsistent damit sammelten Leberzellen ohne MLKL weniger oxidative DNA‑Schäden an, wie eine verringerte Färbung eines Schädigungsmarkers in Zellkernen zeigte. Da langfristige oxidative DNA‑Schäden ein treibender Faktor für krebsauslösende Mutationen sind, trug die Verringerung dieses Stresses wahrscheinlich zum geringeren Tumorbefund bei.
Was das für künftige Behandlungen bedeuten könnte
Zusammen deuten diese Ergebnisse darauf hin, dass MLKL innerhalb von Leberparenchymzellen als stiller Förderer von Leberkrebs im Kontext einer fetten, entzündeten Leber wirkt. Es scheint dies nicht nur durch das Töten von Zellen zu bewirken, sondern auch durch das Stören des mitochondrialen Gleichgewichts, die Verstärkung von oxidativem Stress und das Aufrechterhalten schädlicher Entzündungen und DNA‑Verletzungen. Für Patientinnen und Patienten bedeutet das, dass MLKL ein attraktives Arzneimittelziel sein könnte: Seine Hemmung speziell in Leberzellen könnte helfen, MASH‑bedingten Leberkrebs zu verhindern oder hinauszuzögern. Gleichzeitig muss jede künftige Therapie sehr gezielt sein, denn MLKL erfüllt auch nützliche Funktionen in Immunzellen; das Abschalten dieses Proteins sollte die Leber schützen, ohne die allgemeine Abwehr des Körpers zu schwächen.
Zitation: Imerzoukene, G., Kara-Ali, G.H., Heitz-Marchaland, C. et al. MLKL in liver parenchymal cells promotes liver cancer in murine metabolic dysfunction-associated steatotic liver disease. Cell Death Dis 17, 229 (2026). https://doi.org/10.1038/s41419-026-08458-x
Schlüsselwörter: Fettlebererkrankung, Leberkrebs, Entzündung, oxidativer Stress, MLKL-Protein