Clear Sky Science · de
KDM5B arbeitet mit dem CRL4B‑Komplex zusammen, um die Tumorentstehung von ER+ Brustkrebs durch die Regulierung des Cholesterinstoffwechsels zu fördern
Warum diese Forschung für die Gesundheit im Alltag wichtig ist
Brustkrebs ist die häufigste Krebserkrankung bei Frauen, und viele Tumoren wachsen als Reaktion auf das Hormon Östrogen. Diese Krebsarten teilen sich nicht nur schnell – sie stellen auch um, wie sie Fette wie Cholesterin verwenden. Diese Studie deckt auf, wie ein genregulierendes Protein namens KDM5B mit einem anderen Proteinkomplex, CRL4B, zusammenarbeitet, um das Cholesterin in östrogenrezeptor‑positiven (ER+) Brustkrebszellen zu erhöhen. Wenn man diese verborgene Partnerschaft versteht, hoffen Wissenschaftler, neue Wege zu finden, das Tumorwachstum zu bremsen und vorhandene Behandlungen wie Hormontherapie und cholesterinsenkende Medikamente zu verbessern.
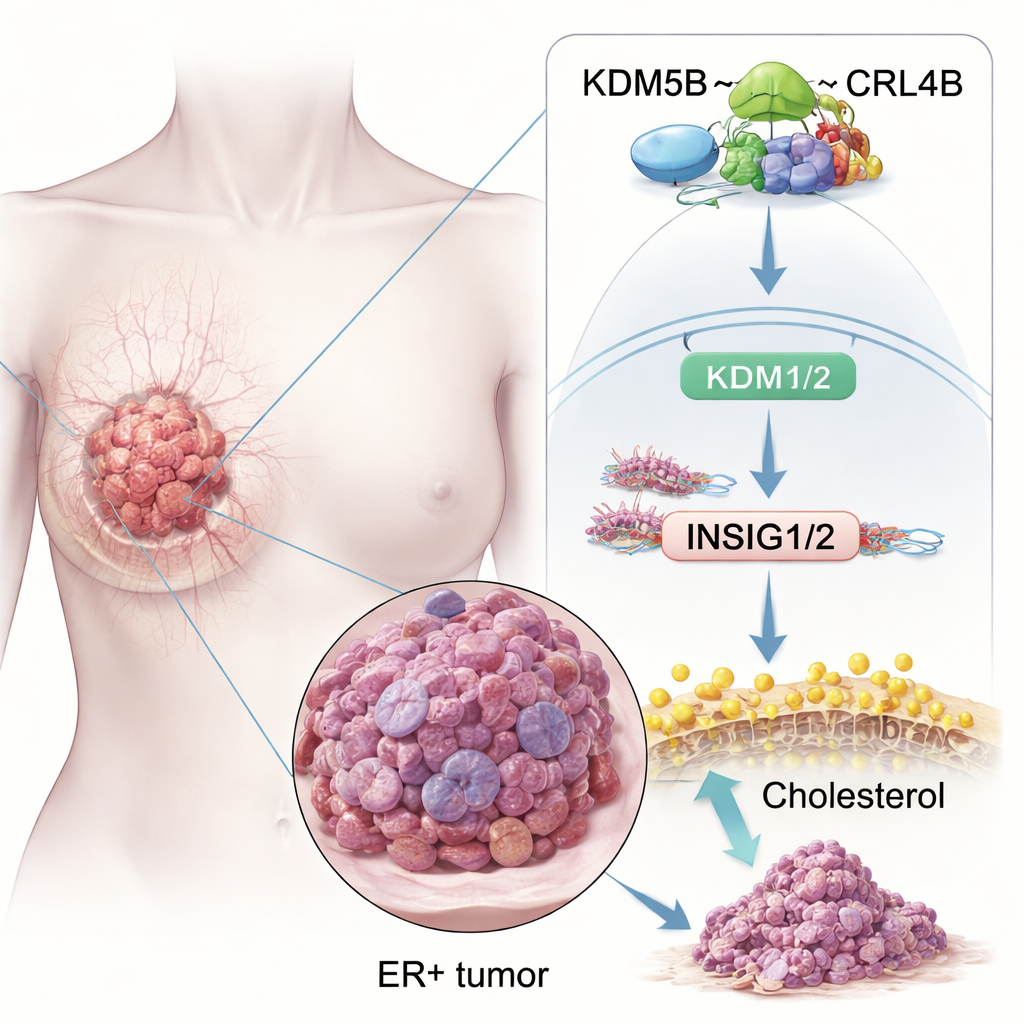
Ein Protein, das das Gleichgewicht zugunsten von Krebs verschiebt
Die Forscher fragten zunächst, ob KDM5B bloß in ER+ Brustkrebserkrankungen vorhanden ist oder ob es diese tatsächlich antreibt. Durch die Analyse großer Krebsdatenbanken und Gewebeproben stellten sie fest, dass die KDM5B‑Werte in Brusttumoren höher sind als im normalen Brustgewebe und besonders hoch in ER+ Tumoren. Patientinnen, deren Tumoren mehr KDM5B produzieren, haben tendenziell eine schlechtere Überlebensrate, selbst wenn sie Standard‑Chemotherapie oder Hormontherapie erhalten. In Zellkulturversuchen führte eine Erhöhung von KDM5B dazu, dass ER+ Brustkrebszellen schneller wuchsen, leichter in das umliegende Gewebe eindrangen und mehr stammzellähnliche Cluster bildeten, die als Ausgangspopulationen für neue Tumoren gelten. Eine Reduktion von KDM5B hatte den gegenteiligen Effekt: Kolonien in der Petrischale und Tumoren in Mäusen wurden kleiner.
Eine mächtige Partnerschaft innerhalb der Krebszellen
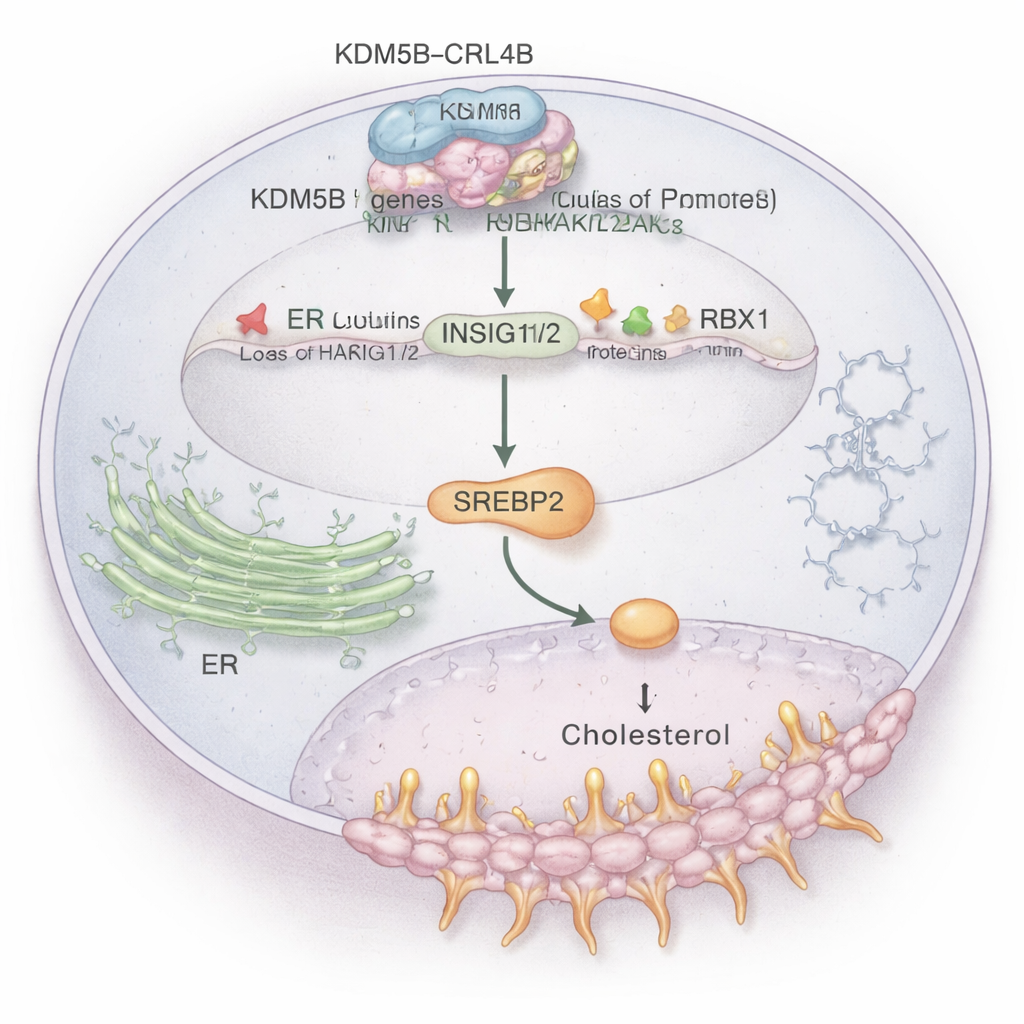
Um zu verstehen, wie KDM5B diese Effekte ausübt, suchte das Team nach seinen Proteinpartnern. Sie entdeckten, dass KDM5B physisch an Teile einer molekularen Maschinerie namens CRL4B‑Komplex bindet, einem Enzymsystem, das Proteine markiert und außerdem die Verpackung der DNA verändert. Detaillierte biochemische Tests zeigten, dass KDM5B direkt mit zwei CRL4B‑Komponenten, CUL4B und DDB1, über spezifische Regionen jedes Proteins interagiert. In ER+ Brustkrebszellen wirkt dieser KDM5B–CRL4B‑Komplex an vielen Genen an deren Ein‑/Ausschaltstellen. Mittels genomweiter Kartierung fanden die Autorinnen und Autoren heraus, dass KDM5B und CUL4B häufig gemeinsam an Promotoren sitzen, wo sie chemische Marker an Histonproteinen – den Spulen, um die die DNA gewickelt ist – verändern, um Gene herunterzuregulieren.
Die Bremsen beim Cholesterin lösen
Unter vielen betroffenen Signalwegen stach der Cholesterinstoffwechsel hervor. Krebszellen benötigen zusätzliches Cholesterin, um Membranen aufzubauen und Stress zu überstehen, und ER+ Tumoren nutzen auch cholesterinabgeleitete Moleküle, um Östrogen nachzuahmen. Die Studie zeigt, dass der KDM5B–CRL4B‑Komplex direkt an die Kontrollregionen zweier wichtiger „Bremsen“-Gene, INSIG1 und INSIG2, bindet. Diese Gene helfen normalerweise, einen zentralen Cholesterinregulator, SREBP2, in Schach zu halten. KDM5B–CRL4B fügt an den Histonen an den INSIG1/2‑Promotoren ein repressorisches Mark (H2AK119ub1) hinzu und entfernt ein aktivierendes Mark (H3K4me3), wodurch diese Gene heruntergeregelt werden. Mit weniger INSIG1/2‑Proteinen wird SREBP2 frei, Cholesterinproduktionsgene zu aktivieren, was den Cholesterinspiegel in ER+ Brustkrebszellen erhöht und ihr invasives Verhalten fördert. Wenn die Forschenden KDM5B oder CRL4B störten, stiegen INSIG1/2, die SREBP2‑Aktivität fiel und der Gesamtcholesterinspiegel in den Zellen nahm ab.
Cholesterin‑Medikamente und Krebs‑Signale kreuzen sich
Die Arbeit verknüpft diesen Weg außerdem mit einem cholesterinabgeleiteten Signal, 27‑Hydroxycholesterin (27‑HC), einem Molekül, das bereits dafür bekannt ist, ER+ Brustkrebs zu fördern. Die Behandlung von ER+ Zellen mit 27‑HC erhöhte KDM5B‑Spiegel und unterdrückte weiter INSIG1/2, was die Zellen zu mehr Wachstum und Invasion trieb. Wichtig ist, dass das Blockieren von KDM5B oder CRL4B diese aggressiven Effekte abschwächte, was darauf hindeutet, dass 27‑HC teilweise durch die Einbindung in die KDM5B–CRL4B‑Achse wirkt. Separat zeigten die Forschenden, dass Simvastatin, ein weit verbreiteter cholesterinsenkender Statin, das Wachstum von Brustkrebszellen verlangsamte und in Kombination mit einem KDM5B‑Inhibitor die Anti‑Tumor‑Wirkung stärker war. Das deutet darauf hin, dass die Kombination von Medikamenten, die die Cholesterinproduktion hemmen, mit Wirkstoffen, die KDM5B′s genkontrollierende Aktivität angreifen, eine vielversprechende therapeutische Strategie sein könnte.
Was das für Patientinnen und zukünftige Behandlungen bedeutet
Diese Studie enthüllt eine neue Abfolge von Ereignissen in ER+ Brustkrebszellen: Ein cholesterinbezogenes Signal (27‑HC) steigert KDM5B; KDM5B arbeitet mit dem CRL4B‑Komplex zusammen, um INSIG1 und INSIG2 herunterzufahren; dadurch wird SREBP2 entfesselt, die Cholesterinproduktion hochgefahren und Tumoren wachsen, dringen ein und erhalten stammzellähnliche Zellen. Da KDM5B auch in mehreren anderen Krebsarten erhöht ist und mit schlechterem Überleben assoziiert wird, könnte das Blockieren dieses Proteins – oder das Wiederherstellen der INSIG1/2‑Bremsen – neue Wege bieten, Tumorwachstum zu kontrollieren. Obwohl weitere Forschung nötig ist, bevor dies in die Routineversorgung übersetzt werden kann, unterstreichen die Ergebnisse, wie eng das Krebsverhalten mit alltäglichen Molekülen wie Cholesterin verknüpft ist und wie vorhandene Medikamente wie Statine eines Tages mit epigenetischen Therapien kombiniert werden könnten, um die Behandlungsergebnisse zu verbessern.
Zitation: Yang, Y., Gao, T., Yuan, B. et al. KDM5B cooperates with CRL4B complex to promote the tumorigenesis of ER+ breast cancer via regulating cholesterol metabolism. Cell Death Dis 17, 207 (2026). https://doi.org/10.1038/s41419-026-08438-1
Schlüsselwörter: ER‑positiver Brustkrebs, Cholesterinstoffwechsel, KDM5B, INSIG1/INSIG2, epigenetische Therapie