Clear Sky Science · de
Mutantes TDP-43 verursacht Beeinträchtigungen im axonalen Transport und in der Glykolyse in einem aus murinen Stammzellen abgeleiteten Motorneuron-Modell der amyotrophen Lateralsklerose (ALS)
Warum diese Forschung für Menschen mit ALS wichtig ist
Die amyotrophe Lateralsklerose (ALS) ist eine tödliche Krankheit, die Menschen schrittweise lähmt, indem sie die Nervenzellen tötet, die Muskeln steuern. Bei den meisten Patientinnen und Patienten findet sich Schädigung, die ein Protein namens TDP-43 betrifft, doch Wissenschaftlerinnen und Wissenschaftler verstehen noch nicht vollständig, wie diese Schädigung Motorneurone tatsächlich schädigt. Diese Studie verwendet ein sorgfältig entwickeltes Maus-Stammzellmodell, um einige der frühesten Probleme zu identifizieren, die durch eine krankheitsassoziierte Variante von TDP-43 verursacht werden, und liefert Hinweise, die künftige Behandlungsansätze informieren könnten.
Motorneurone in der Petrischale erzeugen
Um ALS in einer kontrollierten Umgebung zu untersuchen, begannen die Forschenden mit murinen embryonalen Stammzellen und lenkten sie in Richtung Motorneurone — die langen, kabelartigen Zellen, die Signale vom Rückenmark zu den Muskeln senden. Sie fügten diesen Zellen eine einzelne zusätzliche Kopie des humanen TDP-43-Gens ein, entweder in seiner normalen Form oder mit einer spezifischen, mit ALS assoziierten Mutation namens M337V. Ein fluoreszierender Tag erlaubte dem Team, das humane Protein innerhalb der Zellen zu verfolgen. Am Tag 20 der Zellkultur waren sowohl die normalen als auch die mutierten Zellen zu Motorneuronen gereift, die typische Marker exprimierten, verzweigte Netzwerke bildeten und synapsenähnliche Verbindungen ausbildeten — ein Verhalten, das Neuronen im Nervensystem sehr ähnlich ist. 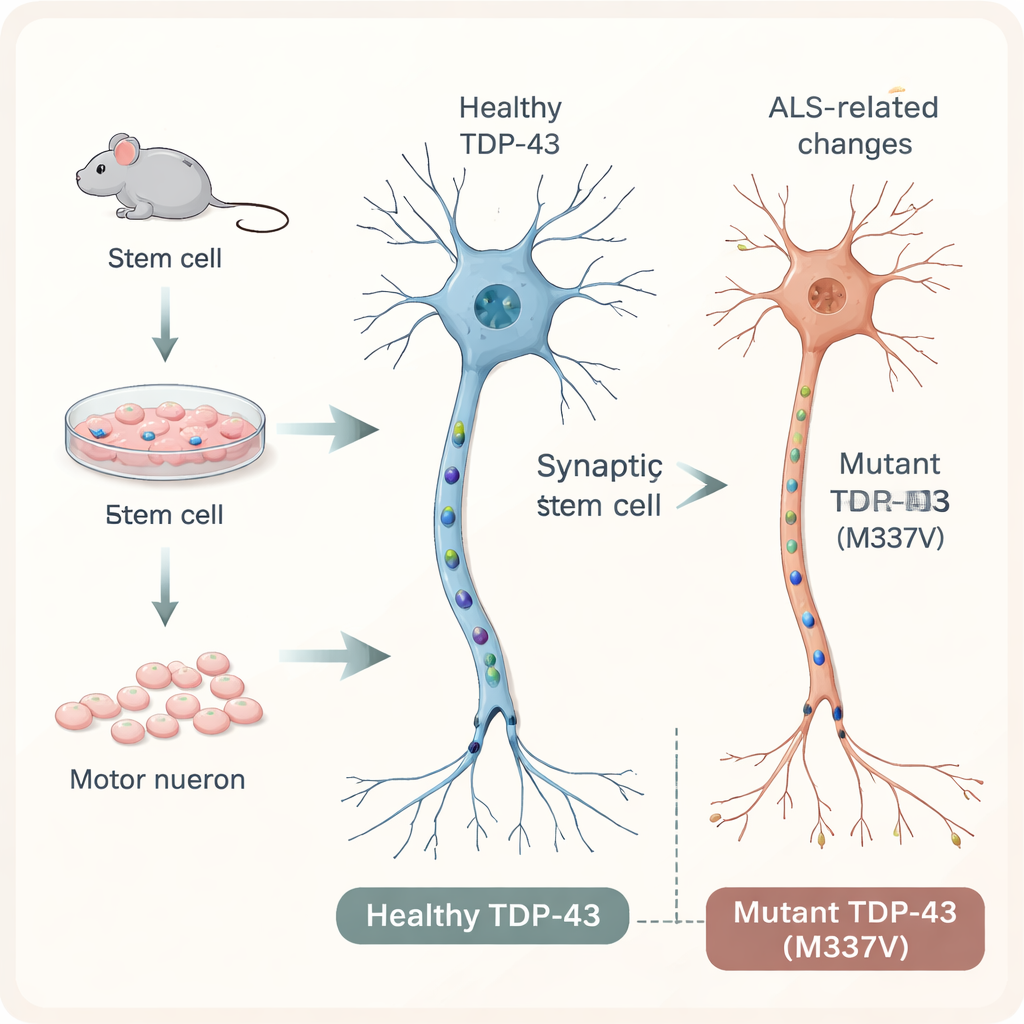
Verborgene Schäden ohne sichtbare Proteinaggregate
Bei Menschen mit ALS verlagert sich TDP-43 oft aus seiner üblichen Position im Zellkern in das zytoplasmatische Kompartiment und bildet Aggregate — ein klassisches pathologisches Merkmal unter dem Mikroskop. Überraschenderweise zeigte das mutierte TDP-43 in diesem Stammzellmodell im Vergleich zur normalen Variante keine vermehrte Fehlverteilung oder Aggregation. Der Großteil des Proteins blieb an seinem vorgesehenen Ort. Dennoch waren die Neurone deutlich weniger gesund: Kulturen mit dem mutierten Protein wiesen weniger Zellkörper, kleinere Netze von Nervenfasern und eine insgesamt reduzierte Lebensfähigkeit auf. Das deutet darauf hin, dass schwerwiegende Schäden an Motorneuronen bereits vor — oder sogar ohne — die dramatischen Proteinaggregate auftreten können, die in Gehirn und Rückenmark von Patientinnen und Patienten sichtbar sind.
Staus entlang der neuronalen „Autobahnen”
Motorneurone sind auf schnelle, effiziente Transportsysteme angewiesen, die lebenswichtige Fracht entlang ihrer langen Axone hinauf und hinunter bewegen. Mithilfe mikrofluidischer Geräte, die Axone in winzigen Kanälen isolieren, verfolgte das Team die Bewegung von Signalpaketen (Endosomen) und energieerzeugenden Mitochondrien in lebenden Zellen. In Neuronen mit mutiertem TDP-43 bewegten sich diese Lasten zwar meist in die richtigen Richtungen, aber langsamer. Signal-endosomen zeigten verringerte Geschwindigkeiten beim Rücktransport zum Zellkörper, und Mitochondrien bewegten sich in beiden Richtungen träger und verbrachten mehr Zeit angehalten. Wichtig ist, dass die grundlegende Maschinerie, die diese Bewegung antreibt — die Motorproteine, die entlang interner Schienen „laufen“ — mengenmäßig offenbar unverändert blieb, was darauf hindeutet, dass das Problem eher in der Funktion dieser Maschinerie liegt als in ihrer Menge.
Energieengpässe bei der Zuckerverbrennung, nicht bei den Mitochondrien
Da axonaler Transport energieintensiv ist, untersuchten die Forschenden, wie diese Neurone Energie erzeugen und nutzen. Sie maßen zwei Hauptquellen: mitochondriale Atmung, die Brennstoff unter Verwendung von Sauerstoff verbrennt, und Glykolyse, die Zucker im umgebenden Medium abbaut. Mitochondrien erschienen in Anzahl, Form und Membranpotenzial normal, und ihre Gesamtfähigkeit zur Energieproduktion schien in den mutierten Zellen unverändert. Im Gegensatz dazu zeigten die Neurone mit mutantem TDP-43 einen klaren Rückgang der basalen Glykolyse. Frühere Arbeiten haben gezeigt, dass lokale Glykolyse entlang der Axone „an Bord“ verfügbare Energie für den schnellen Transport von Vesikeln bereitstellen kann. Die hier beobachtete reduzierte Zuckerverbrennung könnte daher zur verlangsamten Bewegung der Fracht beitragen und setzt die bereits anfälligen Motorneurone weiter unter Stress.
Was das für zukünftige ALS-Therapien bedeutet
Insgesamt zeigt die Studie, dass bereits geringe Mengen an ALS-assoziiertem mutiertem TDP-43 ausreichen, um Motorneurone anfälliger zu machen, den Transport lebenswichtiger Fracht entlang ihrer Axone zu verlangsamen und ihre Fähigkeit, Energie aus Zucker zu gewinnen, zu dämpfen — und das alles ohne die offensichtlichen Proteinaggregate, nach denen Pathologen normalerweise suchen. Für Nicht-Spezialisten lautet die zentrale Botschaft, dass frühe, subtile Veränderungen im zellulären „Verkehrsfluss“ und im Energieverbrauch den Boden für spätere, dramatischere Schäden bei ALS bereiten können. Dies macht den axonalen Transport und die zellulären Energiewege, insbesondere die Glykolyse, zu vielversprechenden Zielen für Therapien, die darauf abzielen, Motorneurone zu schützen, bevor irreversible Degeneration eintritt. 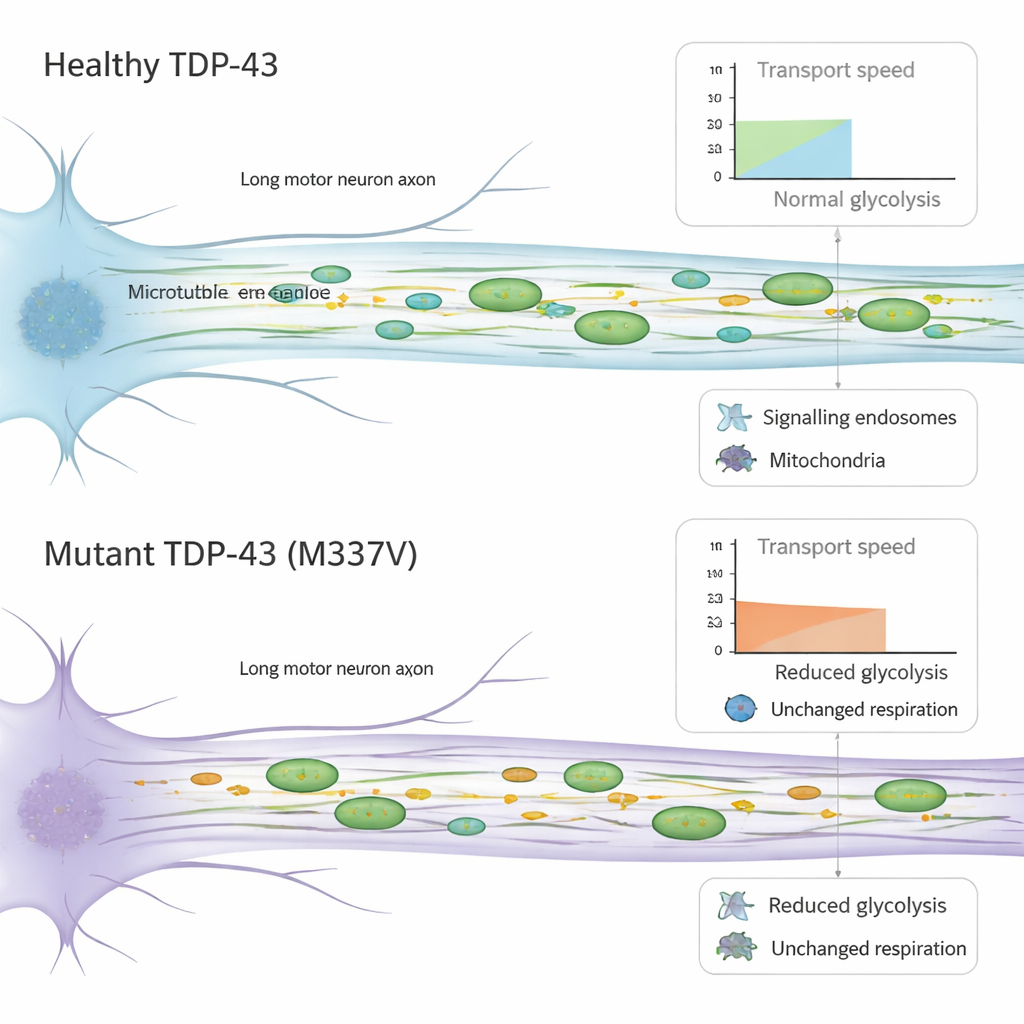
Zitation: Carroll, E., Scaber, J., Pasniceanu, IS. et al. Mutant TDP-43 drives impairments in axonal transport and glycolysis in a mouse stem-cell-derived motor neuron model of amyotrophic lateral sclerosis (ALS). Cell Death Dis 17, 193 (2026). https://doi.org/10.1038/s41419-026-08437-2
Schlüsselwörter: amyotrophe Lateralsklerose, TDP-43, Motorneurone, axonaler Transport, zelluläre Energie