Clear Sky Science · de
LAP2α treibt Brusttumorentstehung voran, indem es Replikationsstress mindert
Warum das für Brustkrebs wichtig ist
Brusttumore wachsen teilweise, weil Krebszellen Wege finden, dem ständigen Stress beim Kopieren ihrer DNA zu trotzen. Diese Studie zeigt, wie ein weniger bekanntes Protein, genannt LAP2α, Brustkrebszellen dabei hilft, ihre DNA während der Replikation zu schützen, wodurch Tumorwachstum gefördert und die Behandlung erschwert wird. Das Verständnis dieses verborgenen Unterstützungssystems weist auf neue Möglichkeiten hin, Tumoren zu schwächen und die Wirkung vorhandener Medikamente zu verbessern.
Ein verborgener Helfer im Zellkern von Tumorzellen
Jedes Mal, wenn eine Zelle sich teilt, muss sie ihre DNA genau kopieren. In Krebszellen ist dieser Prozess besonders belastet: Wachstumssignale stehen dauerhaft auf „ein“ und die Replikationsmaschinerie wird bis an ihre Grenzen getrieben. Dieser Druck, bekannt als Replikationsstress, hinterlässt Bereiche einzelsträngiger DNA, die schnell von einem Protein komplex namens RPA überzogen und stabilisiert werden müssen. Die Autoren hatten zuvor gezeigt, dass LAP2α, ein Protein, das mit dem inneren Gerüst des Zellkerns verbunden ist, RPA binden und ihm helfen kann, beschädigte DNA zu erreichen. In der vorliegenden Arbeit fragten sie, ob diese LAP2α–RPA-Partnerschaft tatsächlich die Entwicklung von Brusttumoren antreibt und die Reaktion auf Therapien beeinflusst.
Hohe LAP2α-Werte kennzeichnen aggressivere Brusttumore
Durch die Analyse großer öffentlicher Expressionsdatensätze und die Untersuchung von Tumorproben unter dem Mikroskop zeigten die Forschenden, dass LAP2α in Brustkrebsgewebe durchgehend höher ist als im umliegenden normalen Brustgewebe. Sein Gehalt steigt mit dem Tumorgrad, das heißt: stärker entartete und aggressivere Tumoren weisen tendenziell mehr LAP2α auf. Dieses Muster zeigte sich in mehreren wichtigen Brustkrebs-Subtypen, einschließlich hormonrezeptorpositiver, HER2-angereicherter und triple-negativer Tumoren. Für Patientinnen wichtig: Tumoren mit hohem LAP2α gingen mit schlechterem Überleben einher. Im Gegensatz dazu zeigten die Kernkomponenten von RPA selbst keine ähnlichen Veränderungen oder klare Zusammenhänge mit dem Ausgang, was darauf hindeutet, dass die verstärkte Funktion von LAP2α – und nicht einfach eine höhere RPA-Menge – Krebszellen besser mit Replikationsstress umgehen lässt.
LAP2α ausschalten verlangsamt Tumoren und legt Schwachstellen frei
Um über Korrelationen hinauszugehen, verwendete das Team Mausmodelle für Brustkrebs, in denen LAP2α selektiv entfernt werden konnte. Nachdem sich Mammatumoren gebildet hatten, führte die genetische Deletion von LAP2α zu langsamerem Tumorwachstum und verlängertem Überleben der Tiere. Tumorzellen ohne LAP2α teilten sich weniger und zeigten mehr Anzeichen von DNA-Schäden, sichtbar durch erhöhte Färbung für Marker von DNA-Brüchen und geringere RPA-Bedeckung der DNA. Als diese LAP2α-defizienten Tumorzellen in neue Mäuse transplantiert wurden, bildeten sie erneut kleinere Tumoren und zeigten eine erhöhte Empfindlichkeit gegenüber DNA-schädigender Chemotherapie, einschließlich des Platinumswirkstoffs Cisplatin und eines PARP-Inhibitors. Ähnliche Experimente in humanen Brustkrebszelllinien bestätigten, dass eine Reduktion von LAP2α Zellen gegenüber mehreren genotoxischen Wirkstoffen verwundbarer machte, während die Wiederherstellung normalen LAP2α – nicht jedoch einer Mutante, die RPA nicht binden kann – sowohl den DNA-Schutz als auch die Arzneimittelempfindlichkeit wiederherstellte.
Wie LAP2α fragile DNA-Stränge schützt
Um den Mechanismus zu klären, rekonstruierten die Forschenden die DNA-Bindungsschritte in Reagenzglasversuchen. Sie mischten gereinigtes RPA, einzelsträngige DNA und entweder normales LAP2α oder eine Variante, die nicht mit RPA interagiert. Sie stellten fest, dass LAP2α direkt die Effizienz erhöhte, mit der RPA einzelsträngige DNA überzog, und RPA dabei half, diese empfindlichen Bereiche zu strecken und zu stabilisieren – ähnlich einem Beladehelfer oder Chaperon. Sobald DNA vorhanden war, löste sich RPA tendenziell von LAP2α und bindete vollständig an den Einzelstrang, was zeigt, dass LAP2α nicht Teil des finalen Schutzmantels ist, sondern RPA an die DNA übergibt. Ohne eine funktionale LAP2α–RPA-Interaktion kollabierten mehr Replikationsgabeln, DNA-Brüche häuften sich und Krebszellen starben häufiger, besonders wenn durch Chemotherapie zusätzliche Schäden induziert wurden.
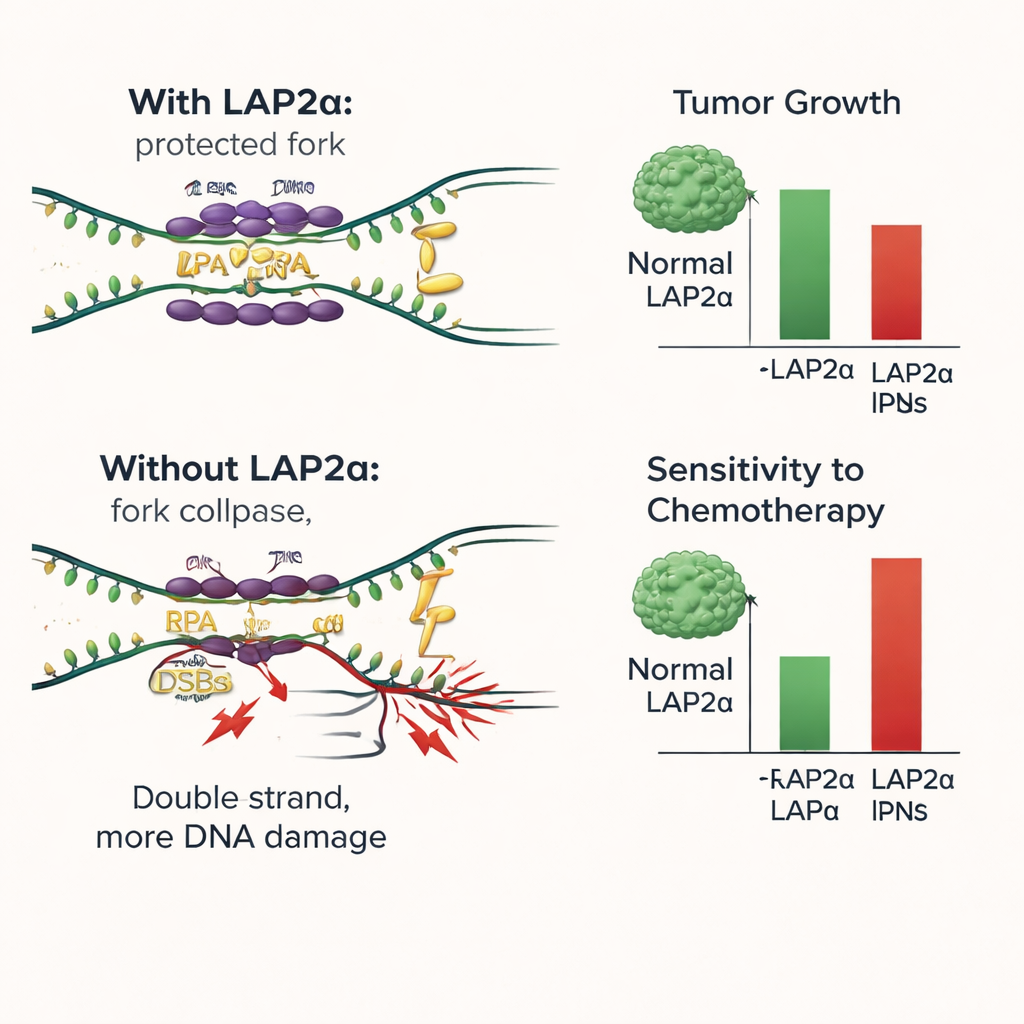
Was das für die künftige Behandlung von Brustkrebs bedeutet
Kurz gesagt zeigt diese Studie, dass LAP2α als eine Art Bühnenhelfer fungiert, der Brustkrebszellen dabei unterstützt, die Belastung beim Kopieren ihrer DNA zu überstehen. Indem es RPA effizient auf verletzliche DNA lädt, verringert LAP2α Schäden und unterstützt weiteres Tumorwachstum. Das Entfernen oder Deaktivieren von LAP2α kippt das Gleichgewicht: DNA-Schäden häufen sich, Zellen hören auf, sich zu teilen, und konventionelle Medikamente, die DNA angreifen, werden wirksamer. Diese Ergebnisse legen nahe, dass LAP2α als Marker für schlechte Prognose und als neues therapeutisches Ziel dienen könnte. Wirkstoffe, die LAP2α oder seine Bindung an RPA blockieren, könnten vorhandene Therapien wie Platinpräparate und PARP-Inhibitoren besonders in Tumoren, die stark auf dieses Stresspuffer-System angewiesen sind, wirksamer machen.
Zitation: Ma, Y., Qin, Y., Bao, P. et al. LAP2α drives breast tumorigenesis by mitigating replication stress. Cell Death Dis 17, 201 (2026). https://doi.org/10.1038/s41419-026-08433-6
Schlüsselwörter: Brustkrebs, DNA-Replikationsstress, LAP2 alpha, Replikationsprotein A, Empfindlichkeit gegenüber Chemotherapie