Clear Sky Science · de
Abweichende Aufrechterhaltung des Entwicklungs‑Transkriptionsfaktors PAX6 fördert neuronalen Zelltod über JNK3‑Signalgebung
Warum diese Forschung für das Sehen wichtig ist
Glaukom ist eine der Hauptursachen für dauerhafte Erblindung, hauptsächlich weil die Nervenzellen, die visuelle Informationen vom Auge zum Gehirn leiten, allmählich absterben. Viele Behandlungen senken den Augeninnendruck, aber Patientinnen und Patienten können trotz guter Druckkontrolle weiterhin das Sehvermögen verlieren. Diese Studie geht einer tieferen Frage nach: Was veranlasst diese retinalen Nervenzellen, bei Stress den „Entscheid“ zu sterben, und können wir diese Entscheidung auf Ebene der Genregulation im Zellkern abschalten?
Eine gestresste Netzhaut unter Beschuss
Kernproblem beim Glaukom und verwandten Augenerkrankungen ist der langsame Verlust retinaler Ganglienzellen (RGCs), der Ausgangsneurone des Auges. Diese Zellen sind gegenüber vielen Stressarten verwundbar, darunter toxische Mengen des Neurotransmitters Glutamat, das NMDA‑Rezeptoren überaktiviert und schädliche Kalziumüberladung auslöst. Die Forschenden nutzten ein etabliertes Mausmodell, bei dem eine kleine Menge NMDA ins Auge injiziert wird und selektiv RGCs schädigt, während andere Netzhautschichten weitgehend intakt bleiben. Sie bestätigten, dass diese Behandlung den Augeninnendruck nicht veränderte, wohl aber typische Merkmale des programmierten Zelltods in RGCs auslöste, wie die Freisetzung von Cytochrom c aus Mitochondrien und das Auftreten TUNEL‑positiver Zellkerne.
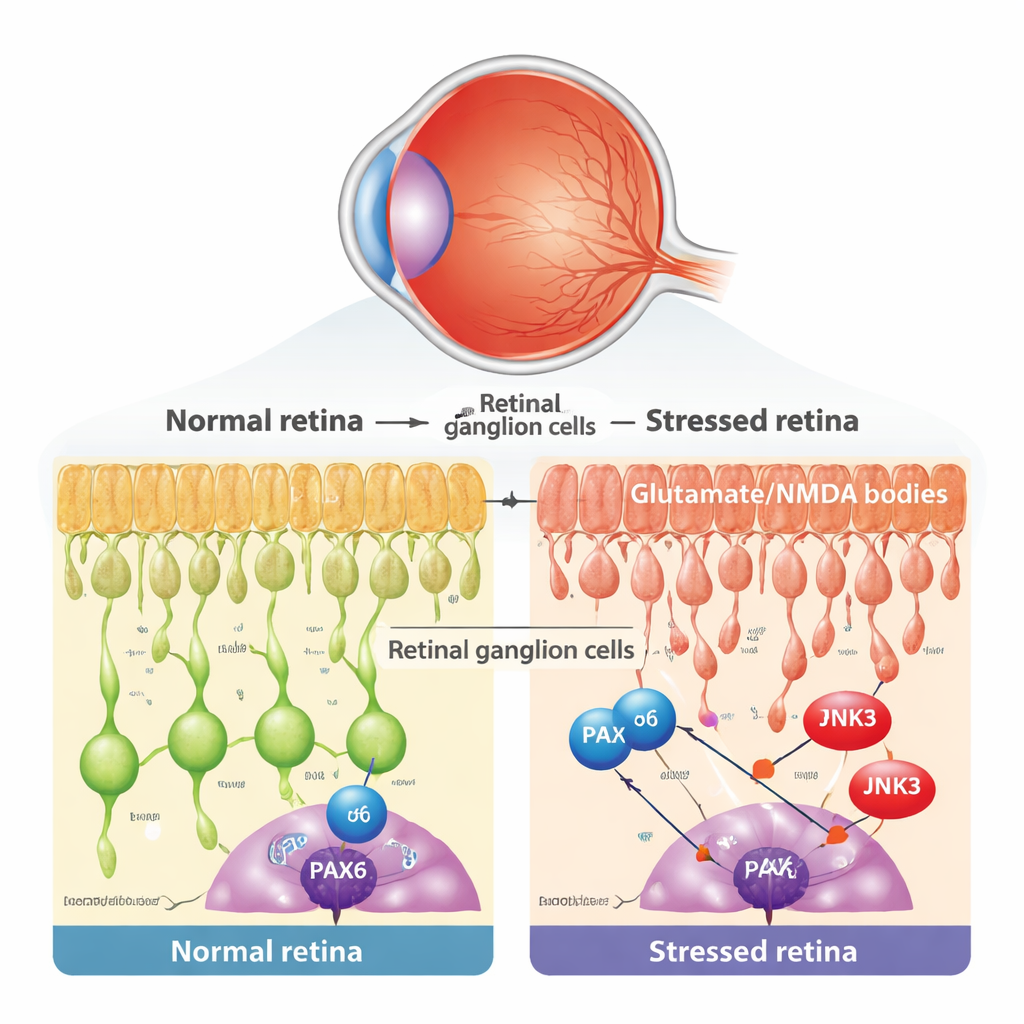
Ein Entwicklungsgen, das sich weigert, in Rente zu gehen
Während der frühen Entwicklung wirkt ein Genregulator namens PAX6 als Masterarchitekt des Auges und steuert, wie verschiedene Netzhautzellen entstehen und verschaltet werden. Üblicherweise gilt, dass solche Entwicklungsprogramme im Erwachsenenalter weitgehend abgeschaltet sind. Durch die Reanalyse von Single‑Cell‑RNA‑Sequenzierungsdaten aus Maus‑ und Menschenretina fand das Team jedoch, dass PAX6 in reifen RGCs und bestimmten Interneuronen tatsächlich stark und selektiv erhalten bleibt. Mit mikroskopischer Färbung zeigten sie, dass in der Schicht, in der RGCs sitzen, PAX6 überwiegend in Ganglienzellen und nicht in benachbarten Amakrinzellen vorkommt. Das wirft die interessante Möglichkeit auf, dass in der adulten Krankheit ein altes Entwicklungsprogramm zweckentfremdet und zum Treiber der Degeneration wird.
Vom Beschützer zum Vollstrecker: PAX6 wechselt die Rolle
Um zu prüfen, ob PAX6 RGCs unter Stress beim Überleben hilft oder ihr Sterben fördert, verwendeten die Wissenschaftler eine therapeutisch anmutende Methode. Sie lieferten einen viralen Vektor, der eine kleine RNA trägt, welche PAX6 im Auge gezielt herunterreguliert, und setzten die Augen dann NMDA aus. Im Vergleich zu Kontrollaugen zeigten PAX6‑depletierte Netzhäute deutlich weniger apoptotische RGCs und weit weniger mitochondriale Schäden, was darauf hindeutet, dass PAX6 für den voll ausgeprägten Zelltod in diesem Modell erforderlich ist. Eine genomweite RNA‑Sequenzierung ergab, dass viele pro‑apoptotische Gene, insbesondere solche, die an mitochondrialem Schaden und Caspase‑Aktivierung beteiligt sind, durch NMDA in normalen Mäusen stark induziert wurden, während diese Induktion bei PAX6‑Silencing abgeschwächt war. Anders gesagt: PAX6 hilft, ein Netzwerk von Genen zu aktivieren, das RGCs über die Kante treibt.
Die Stresskinase, die den PAX6‑Schalter umlegt
Wie aktiviert Stress PAX6, ohne dessen Menge zu erhöhen? Das Team konzentrierte sich auf JNK3, ein stressresponsives Enzym, das vorwiegend in Neuronen vorkommt. Unter NMDA‑Schädigung wanderte JNK3 in den Zellkern der RGCs und ging dort eine physische Wechselwirkung mit PAX6 ein. Biochemische In‑vitro‑Experimente mit gereinigten Proteinen zeigten, dass JNK3 PAX6 direkt phosphorylieren kann, und diese Reaktion wurde durch einen JNK‑Inhibitor blockiert. In Mäusen ohne das Jnk3‑Gen zeigte NMDA nicht mehr dasselbe Muster an PAX6‑Phosphorylierung. Chromatin‑Kartierungen (ChIP‑seq) und gezielte DNA‑Bindungsassays offenbarten, dass phosphoryliertes PAX6 unter Stress zusammen mit JNK3 stärker an Kontrollregionen wichtiger pro‑apoptotischer Gene wie Bax und Gadd45a bindet und deren Aktivität erhöht. Wenn entweder PAX6 herunterreguliert oder JNK3 genetisch entfernt wurde, waren diese Bindung und die entsprechende Aktivierung pro‑tödlicher Gene deutlich reduziert.
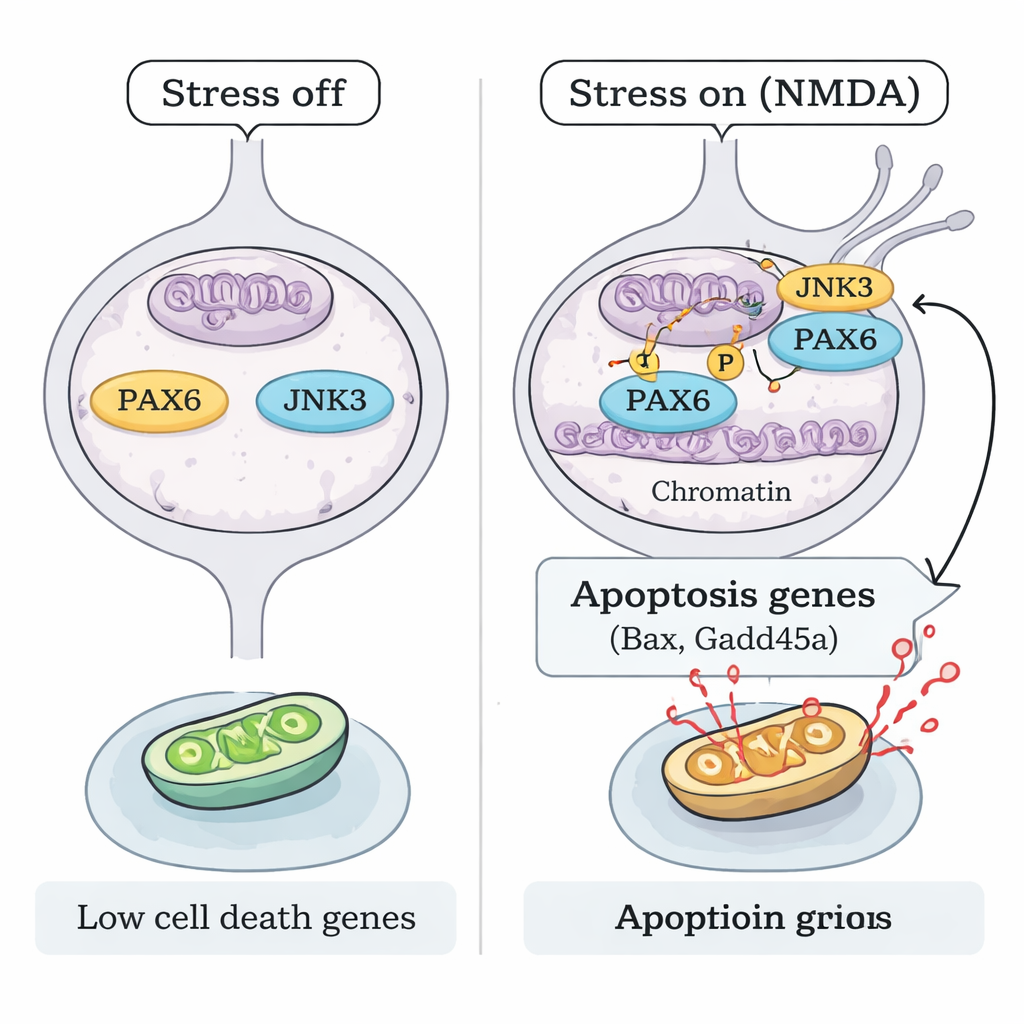
Das Todesprogramm ausschalten, um das Sehen zu schützen
Schließlich fragten die Forschenden, ob das Blockieren dieser JNK3–PAX6‑Achse ausreicht, um sehkritische Zellen zu schützen. Sowohl in PAX6‑Knockdown‑Mäusen als auch in JNK3‑defizienten Mäusen blieben RGCs nach NMDA‑Exposition deutlich besser erhalten, mit weniger sterbenden Zellen und einer gesünderen Netzhautstruktur. Das führt zu einem klaren mechanistischen Modell: Unter exzitotoxischem Stress phosphoryliert JNK3 das persistent exprimierte PAX6 und verwandelt es vom Entwicklungsbauer in einen starken Aktivator eines Zelltod‑Genprogramms in adulten RGCs. Das Unterbrechen dieser Verbindung — durch Silencing von PAX6 oder Deaktivierung von JNK3 — erhält viele dieser Neurone am Leben. Für Patientinnen und Patienten deutet diese Arbeit darauf hin, dass zukünftige Glaukomtherapien über die Senkung des Augeninnendrucks hinausgehen und direkt die genetischen Schalter anvisieren könnten, die darüber entscheiden, ob Netzhautzellen leben oder sterben.
Zitation: Kim, JY., An, MJ., Kim, J. et al. Aberrant maintenance of developmental transcription factor PAX6 promotes neuronal cell death via JNK3 signaling. Cell Death Dis 17, 161 (2026). https://doi.org/10.1038/s41419-026-08417-6
Schlüsselwörter: Glaukom, retinale Ganglienzellen, PAX6, JNK3, Neurodegeneration