Clear Sky Science · de
Dysfunktion GABAerger Interneurone liegt veränderten neuronalen Netzoszillationen zugrunde, die mit epileptiformer Aktivität bei PPT1-defizienten Mäusen verbunden sind
Wenn Gehirnrythmen aus dem Takt geraten
Anfälle sind nicht nur plötzliche Stürme neuronaler Aktivität; sie entwickeln sich oft aus feinen Veränderungen in der Art und Weise, wie Nervenzellen miteinander kommunizieren. Diese Studie untersucht eine seltene kindliche Gehirnerkrankung, die CLN1-Krankheit, und stellt eine einfache Frage mit weitreichenden Implikationen: Was geschieht mit den internen „Taktgebern“ des Gehirns, wenn ein einziges Enzym, PPT1, fehlt? Indem die Forschenden diese Veränderungen bei Mäusen im Zeitverlauf verfolgen, zeigen sie, wie frühe kleine Störungen der Hemmung zu Anfällen und weitreichenden Hirnschäden anwachsen können.
Die Wächter des neuronalen Gleichgewichts
Unser Gehirn beruht auf zwei grundsätzlichen Arten von Nervenzellen. Erregende Zellen, wie Pyramidenneurone im Hippocampus, treiben die Aktivität voran. Hemmende Zellen, sogenannte Interneurone, wirken wie Bremsen, halten diese Aktivität in Schach und formen die elektrischen Rhythmen des Gehirns. Unter ihnen sind zwei wichtige Gruppen parvalbumin-positive (PV+) Interneurone und somatostatin-positive (SST+) Interneurone. Sie helfen, rhythmische Gehirnwellen wie Theta- und Gamma-Oszillationen zu erzeugen und zu koordinieren, die Funktionen wie Lernen und Gedächtnis unterstützen. Bei der CLN1-Krankheit verlieren Kinder das Enzym PPT1, das normalerweise Fettsäuregruppen von Proteinen entfernt. Die Autorinnen und Autoren nutzten ein Mausmodell mit derselben in Patienten vorkommenden Mutation, um zu untersuchen, wie dieser Verlust Interneurone und die von ihnen mitkontrollierten Rhythmen beeinflusst.
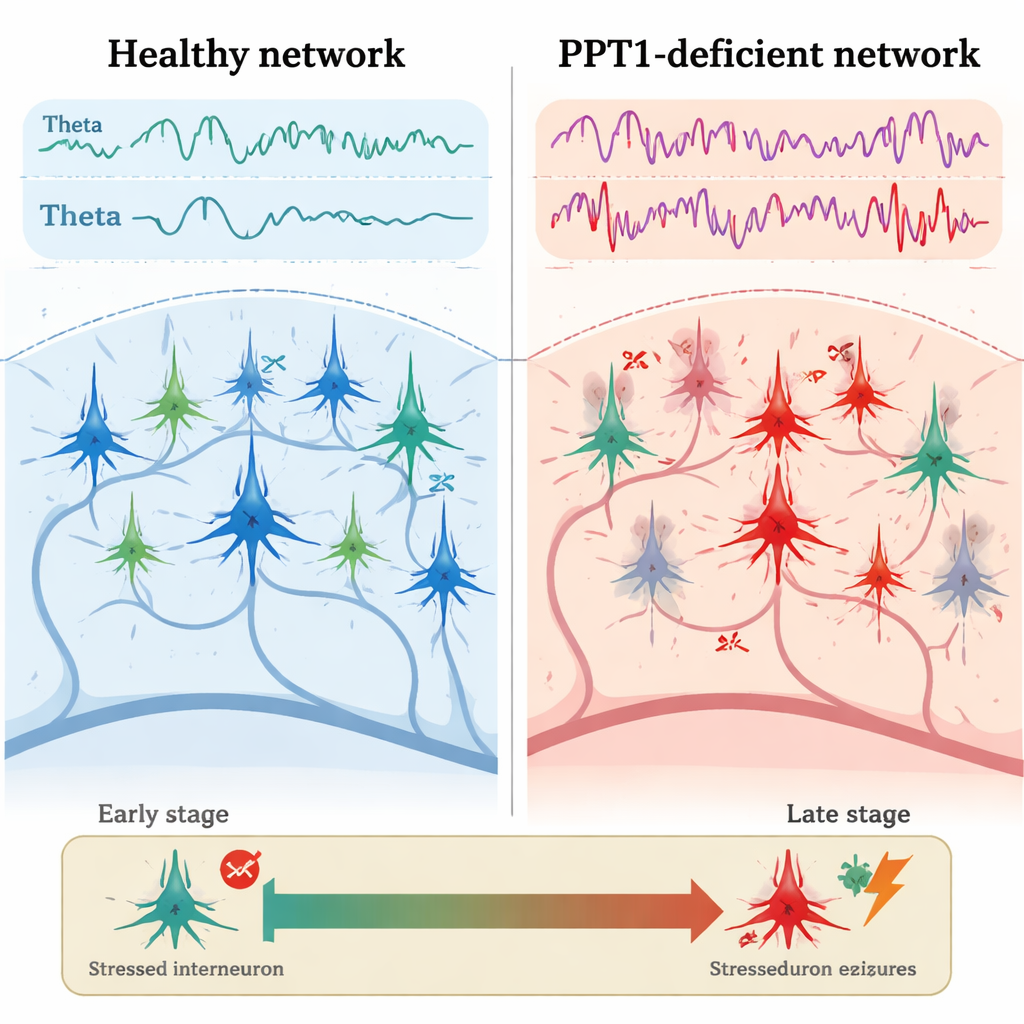
Frühe Risse im hemmenden System
Bei jungen erwachsenen Mutantenmäusen im Alter von etwa drei bis vier Monaten trat das erste klare Problem bei PV+ Interneuronen zutage. Elektrische Aufzeichnungen aus dem Hippocampus zeigten, dass diese hemmenden Zellen seltener feuerten als bei gesunden Mäusen, während benachbarte Pyramidenneurone schneller feuerten und kürzere Pausen zwischen den Spike-Reihen zeigten. Die Mikroskopie offenbarte, dass viele PV+ Interneurone aktiviertes Caspase-3 aufwiesen, einen zentralen Vollstrecker des programmierten Zelltods, obwohl ihre Gesamtzahl noch nicht gesunken war. Gleichzeitig war die Leistung von Theta- und Gamma-Wellen erhöht, und Calciumbildgebung zeigte stärkere Aktivität in Hippocampusneuronen, während sich die Tiere bewegten. Entscheidend war, dass die normale ‚Wechselwirkung‘ zwischen Theta- und Gamma-Rhythmen — bei der langsamere Wellen die schnelleren organisieren — abgeschwächt war, was auf einen frühen Zusammenbruch des feinen Timings neuronaler Netzaktivität hindeutet.
Von gestörten Rhythmen zu Anfallsentladungen
Mit sechs bis sieben Monaten hatte sich das Bild verschlechtert. Viele PV+ Interneurone gingen verloren, und nun zeigten auch SST+ Interneurone Anzeichen von Caspase-3-Aktivierung. Aufzeichnungen aus dem Hippocampus enthüllten spontane epileptoforme Entladungen — kurze, abnorme Aktivitätsausbrüche, die mit Anfällen assoziiert sind. Das Team konzentrierte sich auf hochfrequente ‚Ripples‘, schnelle Oszillationen, die normalerweise bei der Gedächtnisspeicherung helfen. Bei den Mutanten wurden physiologische Ripples (etwa 140–200 Hertz) seltener, hatten aber größere Amplituden, während noch schnellere ‚pathologische‘ Ripples (200–500 Hertz), die eng mit Epilepsie verknüpft sind, stärker und häufiger auftraten. Zusammen deuteten diese Veränderungen auf eine Verschiebung von organisierten, gedächtnisbezogenen Rhythmen hin zu chaotischen, anfallsanfälligen Mustern, während die hemmende Kontrolle versagte.
Neurone verschleißen und Diazepam greift ein
Mit Fortschreiten der Erkrankung begann der Hippocampus selbst zu degenerieren. Calcium-Signale in Neuronen nahmen ab, Golgi-Färbungen zeigten dünnere, weniger verzweigte dendritische Bäume, und es gab weniger kleine Dornen (Spines), an denen Synapsen entstehen. Zellzählungen in wichtigen Hippocampusregionen (CA1 und CA3) bestätigten einen großflächigen Zellverlust, und in elektrischen Aufzeichnungen wurden weniger aktive Einheiten detektiert. Die Forschenden testeten daraufhin Diazepam, ein weit verbreitetes Anti-Anfalls-Medikament, das die Wirkung des inhibitorischen Botenstoffs GABA verstärkt. Bei älteren Mutanten reduzierte Diazepam die Frequenz epileptischer Entladungen und stellte teilweise normalere Oszillationsmuster, einschließlich des Ripple-Verhaltens, wieder her, obwohl es den zugrundeliegenden Rezeptorverlust nicht reparierte. Das deutet darauf hin, dass die Verstärkung verbliebener hemmender Signale das Netzwerk zumindest vorübergehend beruhigen kann.
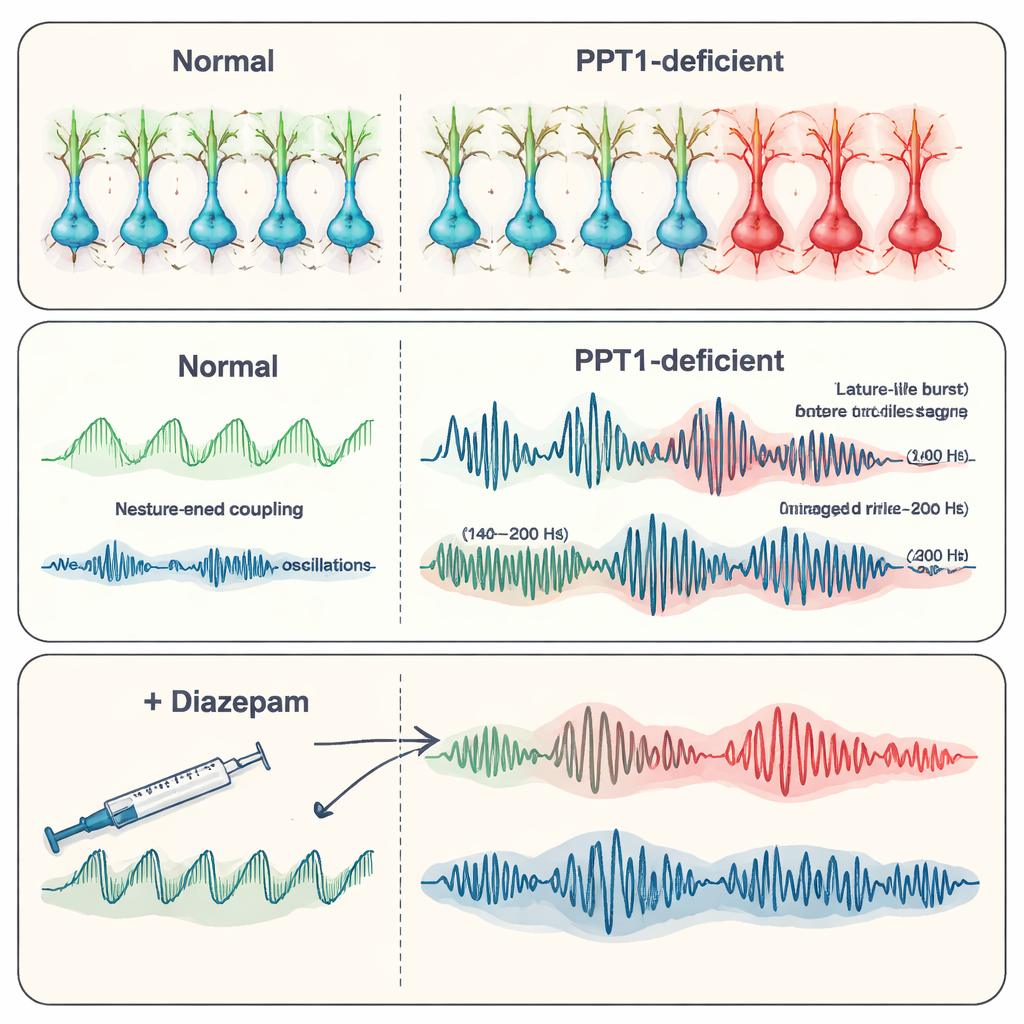
Warum diese Ergebnisse wichtig sind
Für nichtfachliche Leser ist die Kernbotschaft, dass die CLN1-Krankheit nicht nur ein Problem des sich anhäufenden zellulären Abfalls im Gehirn ist. Der Verlust von PPT1 löst eine Kaskade aus: Zuerst geraten spezialisierte hemmende Interneurone unter Stress und beginnen zu versagen, wodurch überaktive Pyramidenneurone entfesselt werden und die Gehirnrythmen verzerrt werden. Im Laufe der Zeit führt dieses Ungleichgewicht zu Anfällen und schließlich zu massivem Verlust von Nervenzellen und Verbindungen. Die Studie weist auf ein Zeitfenster in frühen Krankheitsstadien hin, in dem der Schutz oder die Rettung von PV+ Interneuronen — etwa durch Blockade der Caspase-Aktivierung — spätere Anfälle und Degeneration verhindern könnte. Obwohl Diazepam CLN1 nicht heilen kann, unterstreicht seine Fähigkeit, abnorme Rhythmen in diesem Modell zu dämpfen, die grundsätzliche Idee, dass die Wiederherstellung von Hemmung eine wirkungsvolle Strategie bei Epilepsie und verwandten Hirnerkrankungen sein könnte.
Zitation: Tong, J., Liu, W., Wang, Q. et al. Dysfunction of GABAergic interneurons underlies altered neural network oscillations associated with epileptiform activity in PPT1-deficient mice. Transl Psychiatry 16, 106 (2026). https://doi.org/10.1038/s41398-026-03843-8
Schlüsselwörter: Epilepsie, Interneurone, Hippocampus, Gehirnoszillationen, Lysosomale Speicherkrankheit