Clear Sky Science · de
Steigerung der KLF15-Aktivität in Kardiomyozyten: ein neuartiger Ansatz zur Verhinderung pathologischer Reprogrammierung und Fibrose mittels nuclease-defizientem dCas9VPR
Umschulung des versagenden Herzens
Herzinsuffizienz betrifft Millionen von Menschen und entwickelt sich häufig schleichend nach Jahren von Bluthochdruck oder Klappenerkrankungen. Unter diesen Bedingungen vergrößern sich die Herzmuskelzellen nicht nur, sie schalten auch ein „fötales“ Genprogramm an und das Herz füllt sich mit Narbengewebe. Diese Studie untersucht einen neuen Weg, die körpereigene Gensteuerung des Herzens wieder in Richtung Gesundheit zu lenken – ohne DNA zu schneiden – indem ein schützender Regulator namens KLF15 in Herzmuskelzellen behutsam hochreguliert wird.
Wenn Herz-Zellen ihre Identität verlieren
In einem gesunden Erwachsenenherzen verbrennen Kardiomyozyten – die Herzmuskelzellen – Fette effizient zur Energiegewinnung und halten ein stabiles Genaktivitätsmuster aufrecht. Mithilfe von Einzelzell-RNA-Sequenzierung bei Mäusen unter chronischer Druckbelastung kartierten die Forscher, wie sich einzelne Kardiomyozyten verändern, wenn das Herz von normaler Funktion über Vergrößerung bis hin zum Versagen übergeht. Sie fanden heraus, dass ein Transkriptionsfaktor namens KLF15, der normalerweise Stoffwechsel und Wachstum im Gleichgewicht hält, die stärkste Aktivitätsänderung in kranken Zellen zeigte. Mit zunehmendem Stress fielen KLF15-Spiegel und seine Fähigkeit, fötale und stressbedingte Gene zu unterdrücken, ab. Ähnliche Abnahmen von KLF15 wurden in menschlichen Herzen von Patienten mit dilatativer und hypertropher Kardiomyopathie beobachtet, was darauf hindeutet, dass diese Störung über Arten hinweg erhalten ist.
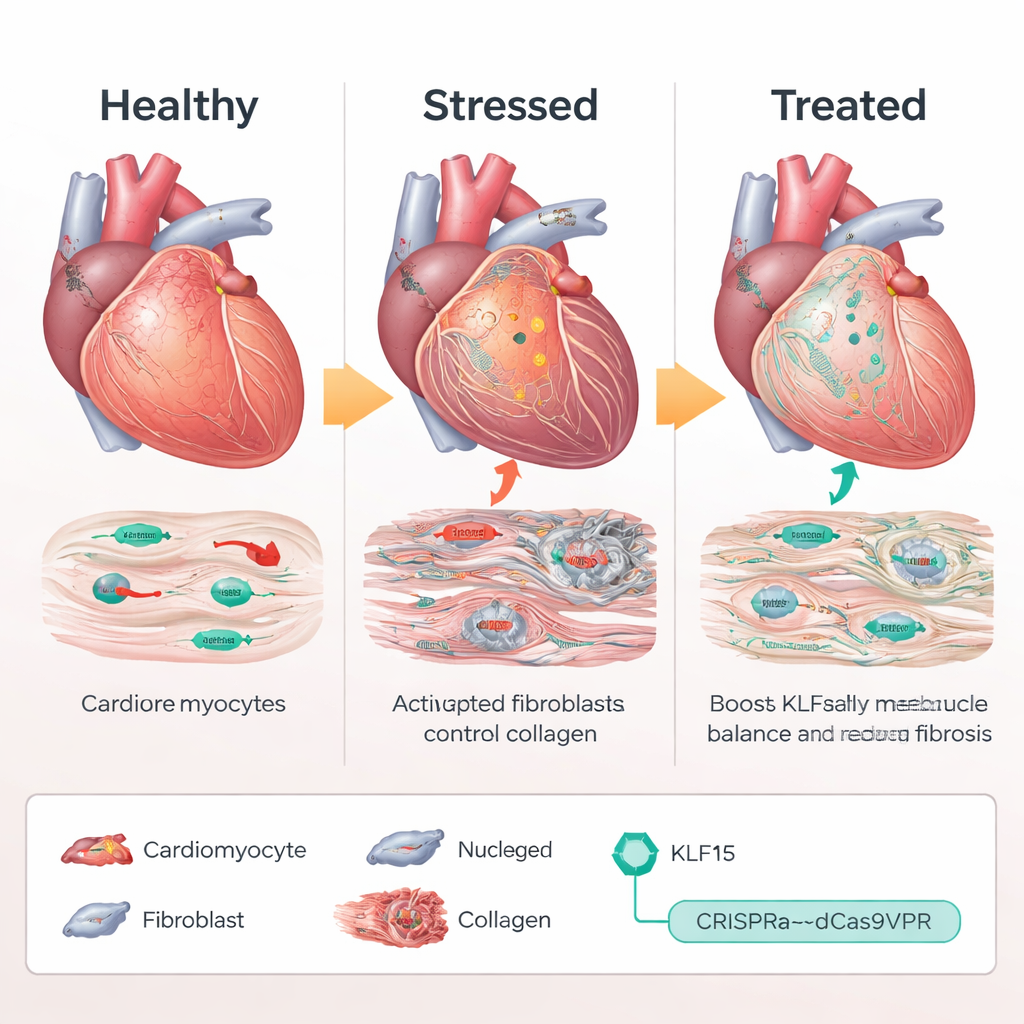
CRISPR als Lautstärkeregler, nicht als Schere
Statt eine zusätzliche Kopie des KLF15-Gens hinzuzufügen oder die DNA zu zerschneiden, verwendete das Team ein CRISPR-basiertes „Aktivierungs“-System, genannt dCas9VPR, das in der Nähe des natürlichen Klf15-Gens bindet und dessen eigene Expression erhöht. In Mäusen, die so konstruiert wurden, dass dieser CRISPR-Aktivator nur in Kardiomyozyten exprimiert wird, lieferten die Wissenschaftler Leit-RNAs mit einem Adeno-assoziierten Virus (AAV9), um den Klf15-Promotor anzusteuern. Unter chronischer Druckbelastung hielten Mäuse, die Klf15-aktivierende Guides erhielten, nahezu normale Klf15-Spiegel aufrecht. Ihre Herzmuskelzellen blieben kleiner, die Pumpfunktion nahm weniger stark ab und die Überlebensrate verbesserte sich im Vergleich zu Kontrolltieren. Auf molekularer Ebene wurden Stress- und Fötal-Gene gedämpft, während Stoffwechsel- und Calcium-handhabende Gene wieder anstiegen, was darauf hinweist, dass das ungesunde Transkriptionsprogramm größtenteils zurückgesetzt wurde.
Verminderung der Narbenbildung durch Zell-zu-Zell-Kommunikation
Herzinsuffizienz wird nicht nur von kranken Muskelzellen vorangetrieben, sondern auch von Fibroblasten, Stütz-Zellen, die Kollagen produzieren und steifes Narbengewebe bilden. Einzelzellanalysen und Gewebebildgebung zeigten, dass die Wiederherstellung von Klf15 in Kardiomyozyten die Aktivierung von Fibroblasten und die Gesamtfibrose reduzierte, obwohl die Gentherapie nie direkt auf Fibroblasten abzielte. Das Team führte diesen Effekt auf ein sezerniertes Protein namens AZGP1 zurück. Wenn Klf15 in Kardiomyozyten erhöht wurde, stiegen Produktion und Freisetzung von AZGP1. Sowohl in Maus-Herzen als auch in menschlichen, aus Stammzellen gewonnenen Herzgeweben dämpfte erhöhtes AZGP1 den TGF-β / SMAD-Signalweg in Fibroblasten – einen zentralen Treiber der Vernarbung – und senkte die Spiegel von Markern wie α-SMA und POSTN. Wichtig ist, dass eine Überexpression von AZGP1 allein in Kardiomyozyten die Muskelzellen nicht umprogrammierte, was zeigt, dass KLF15 primär die Kardiomyozyten direkt schützt und AZGP1 als Botenstoff nutzt, um Fibroblasten zu bremsen.
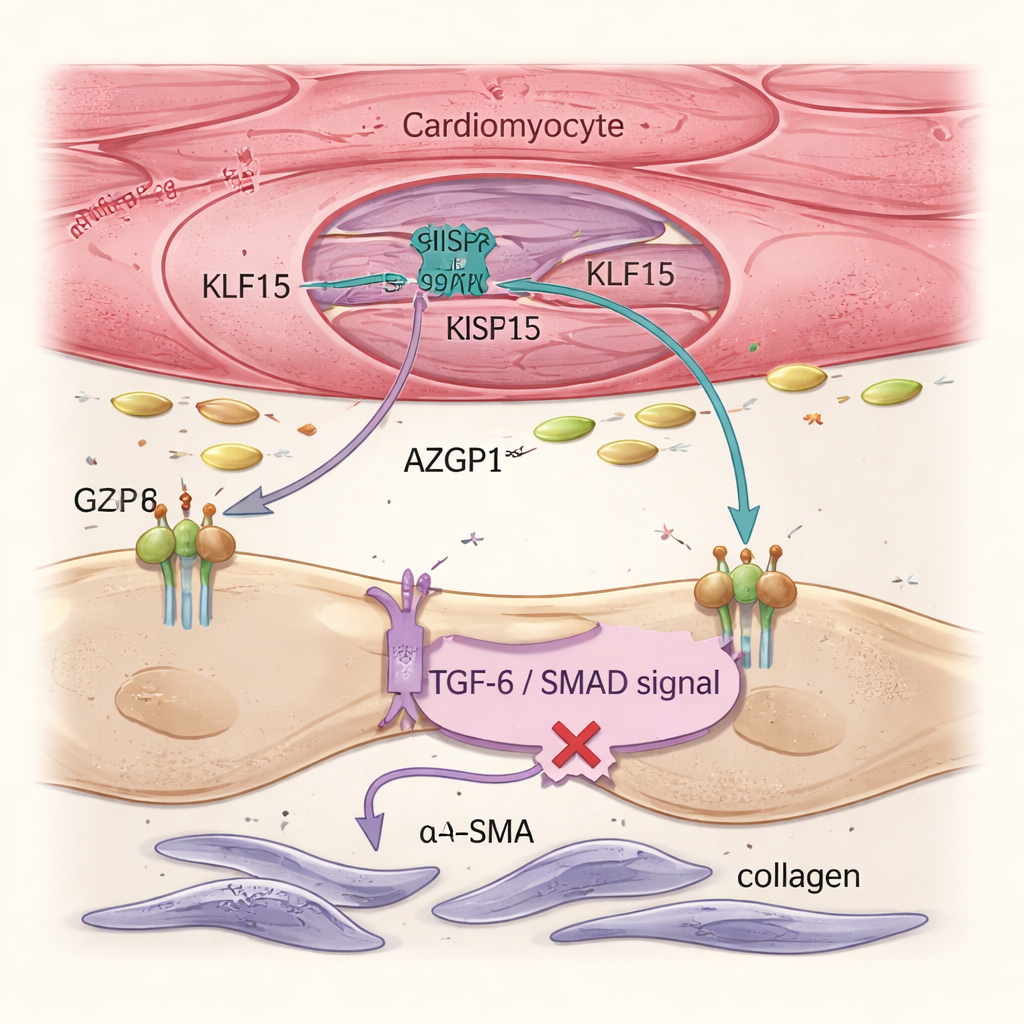
Menschliche Gewebemodelle bestätigen den schützenden Kreislauf
Um zu testen, ob diese Mechanismen auch in menschlichen Zellen gelten, verwendeten die Forscher induzierte pluripotente Stammzell‑abgeleitete Kardiomyozyten, die in dreidimensionalen, konstruierten Herzgeweben kultiviert wurden. Unter mechanischer Belastung, die hohen Blutdruck nachahmt, verloren diese Gewebe KLF15, schalteten Stress- und Fötal-Gene an, versteiften und ihre Kontraktionen schwächten sich – was die Krankheitsmerkmale nachbildete. Die durch CRISPRa vermittelte Wiederherstellung von KLF15 verhinderte diesen Abbau, bewahrte die Kraftentwicklung und verschob die Genexpression zurück in Richtung eines ausgereiften Stoffwechsels und struktureller Merkmale. Detaillierte Experimente zeigten, dass TGF-β1, ein bekannter pro-fibrotischer Signalstoff, KLF15 in menschlichen Kardiomyozyten über den SMAD2/3-Weg reduziert, was erklärt, wie chronischer Stress zu maladaptiver Umbildung führt. Schließlich entwickelte das Team ein kompaktes „Mini“-CRISPRa-System basierend auf einer kleineren Cas9-Variante, das in einen einzelnen AAV9-Vektor passt und von einem kardiomyozytenspezifischen Promotor gesteuert wird. In präzise geschnittenen Scheiben versagender menschlicher Herzgewebe hob dieser Vektor erfolgreich die KLF15-Spiegel an und verbesserte die Kontraktionsleistung über Tage in Kultur.
Ein Fahrplan für sanftere Gentherapie
Für Nicht-Spezialisten ist die Kernbotschaft, dass diese Arbeit zeigt, wie das behutsame Hochregulieren eines einzelnen schützenden Reglers in Herzmuskelzellen sowohl ihre Identität stabilisieren als auch Signale aussenden kann, die die Vernarbung begrenzen. Durch die Verwendung eines CRISPR-basierten Aktivators, der die DNA nicht schneidet, wird das körpereigene Gen feinjustiert, statt ein künstliches einzufügen. Die Studie beschreibt einen TGF-β → KLF15 → AZGP1‑Signalweg, der mechanischen Stress mit schädlicher Umbildung verknüpft, und demonstriert in Mäusen, menschlichen Zellmodellen und menschlichen Herzgewebeschnitten, dass die Wiederherstellung von KLF15 diese Kettenreaktion unterbrechen kann. Obwohl sich der Ansatz noch im präklinischen Stadium befindet, bietet das hier vorgestellte kompakte, kardiomyozyten‑gerichtete CRISPRa‑System einen möglichen Fahrplan zur Behandlung häufiger, nicht‑genetischer Formen der Herzinsuffizienz, indem Genaktivität umprogrammiert statt das Genom umgeschrieben wird.
Zitation: Schoger, E., Kim, R., Bleckwedel, F. et al. Enhancing KLF15 activity in cardiomyocytes: a novel approach to prevent pathological reprogramming and fibrosis via nuclease-deficient dCas9VPR. Sig Transduct Target Ther 11, 76 (2026). https://doi.org/10.1038/s41392-026-02593-9
Schlüsselwörter: Herzinsuffizienz, KLF15, CRISPR-Aktivierung, kardiale Fibrose, AZGP1