Clear Sky Science · de
Parathyroidhormon-verwandtes Protein ist ein therapeutisches Ziel bei idiopathischer Lungenfibrose
Warum Vernarbung der Lunge wichtig ist
Idiopathische Lungenfibrose (IPF) ist eine verheerende Lungenerkrankung, bei der empfindliche Lungenbläschen nach und nach in steifes Narbengewebe umgewandelt werden, sodass jeder Atemzug zur Anstrengung wird. Gegenwärtige Medikamente können dieses Vernarben verlangsamen, aber nicht aufhalten oder rückgängig machen. Diese Studie deckt eine bislang unterschätzte Ursache dieses Prozesses auf: ein kleines, hormonähnliches Protein, das von den Zellen der Atemwegsauskleidung gebildet wird und parathyroidhormon‑verwandtes Protein (PTHrP) genannt wird. Sie zeigt, dass das Blockieren seiner Wirkung einen neuen Behandlungsansatz für IPF eröffnen könnte.
Ein versteckter Botenstoff in den Atemwegen
IPF wurde lange Zeit mit überaktiven Fibroblasten in Verbindung gebracht – Zellen, die normalerweise bei der Gewebereparatur helfen, in dieser Erkrankung jedoch überreagieren und übermäßiges Kollagen einlagern, den Hauptbestandteil von Narben. Viele Studien konzentrierten sich auf Signale von Immunzellen und den tiefsten Lungenbläschenzellen, doch diese Arbeit richtet den Blick höher im Bronchialbaum, auf die Röhren, die die Luft in die Lunge leiten. Durch die Reanalyse großer genetischer Datensätze von Menschen mit und ohne IPF fanden die Forscher, dass ein Gen namens PTHLH, das PTHrP kodiert, in IPF-Lungen konsistent stärker aktiviert war. Mikroskopische Untersuchungen menschlicher Lungenproben bestätigten, dass das PTHrP-Protein bei Patienten mit IPF deutlich vermehrt vorkommt und sich in den bronchialen Epithelzellen konzentriert, die die Atemwege auskleiden.
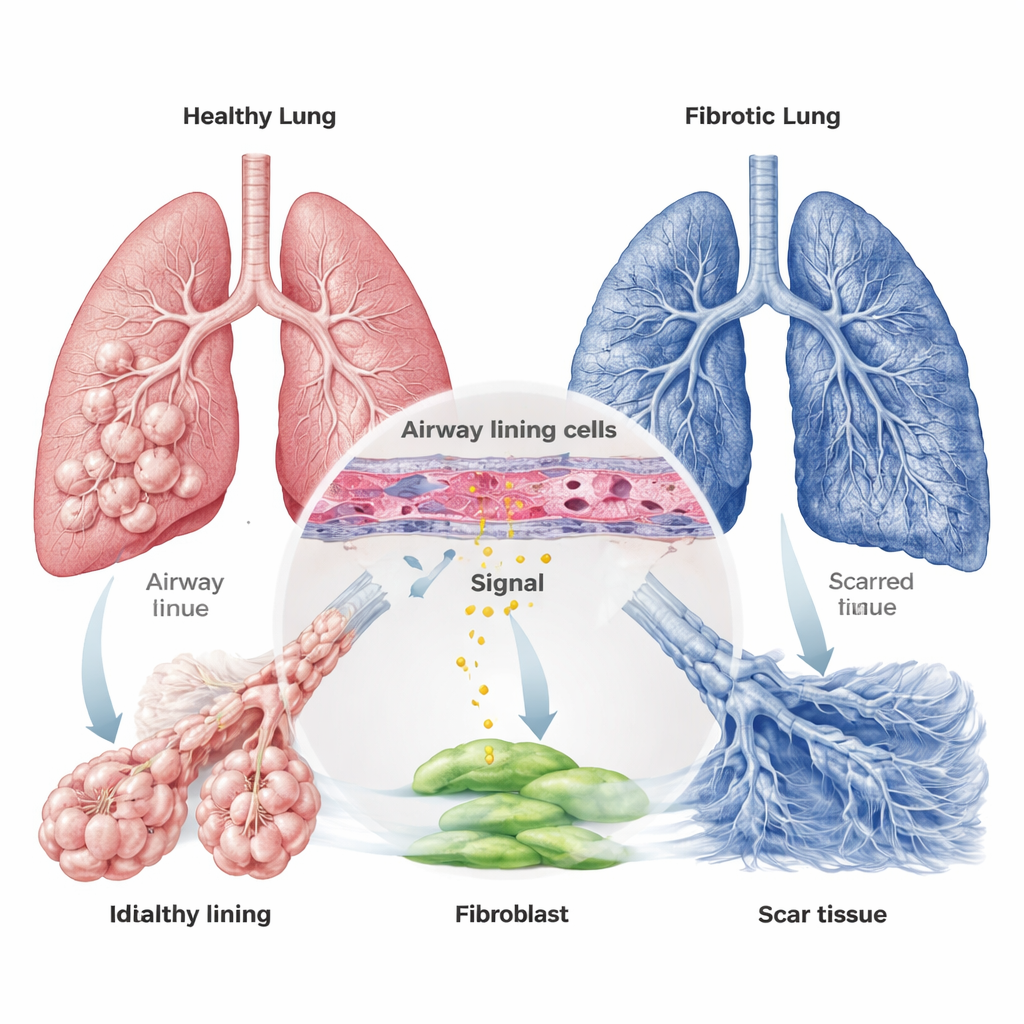
Vom Atemwegssignal zu narbenbildenden Zellen
PTHrP wird als größeres Protein produziert, das in mehrere kleinere Fragmente geschnitten werden kann. Das Team konzentrierte sich auf ein Fragment, PTHrP1‑34, das bekannt dafür ist, einen Rezeptor auf bestimmten Zellen zu aktivieren. Sie zeigten, dass bronchiale Epithelzellen unter Stress – etwa bei Sauerstoffmangel oder nach Exposition gegenüber dem chemotherapieähnlichen Wirkstoff Bleomycin, der häufig zur Modellierung von Lungenverletzung bei Tieren verwendet wird – mehr PTHrP1‑34 in ihre Umgebung freisetzen. Gleichzeitig stellten sie fest, dass der passende Rezeptor, PTH1R, hauptsächlich nicht auf den Zellen der Lungenbläschenoberfläche, sondern auf Fibroblasten und glatten Muskelzellen vorkommt. Das schafft eine Kommunikationslinie: geschädigte Atemwegszellen senden PTHrP1‑34 aus, und benachbarte Fibroblasten sind darauf eingestellt, dieses Signal zu empfangen.
Wie das Signal Fibroblasten aggressiv macht
Im Reagenzglas führte die Zugabe von PTHrP1‑34 zu menschlichen und Maus-Lungenfibroblasten dazu, dass diese einen aggressiveren, narbenbildenden Zustand einnahmen. Sie erhöhten die Produktion von alpha‑glatter Muskelaktin (ein Kennzeichen sogenannter Myofibroblasten) und wichtigen Kollagengenen und zeigten gesteigerte Beweglichkeit – Merkmale, die mit fortschreitender Vernarbung assoziiert sind. Molekulare Untersuchungen zeigten, dass dies über einen spezifischen innerzellulären Weg geschieht: PTHrP1‑34 bindet an PTH1R auf der Fibroblastenoberfläche, steigert den Botenstoff cAMP und aktiviert ein Enzym namens PKA, das dann „Fibrosegene“ im Zellkern einschaltet. Das Blockieren des Rezeptors oder das Hemmen von PKA reduzierte diese Reaktion deutlich. Wichtig ist, dass Epithelzellen der Atemwege und der Lungenbläschen nicht auf dieselbe Weise reagierten, was hervorhebt, dass dieses Hormonfragment sehr selektiv auf Fibroblasten wirkt.
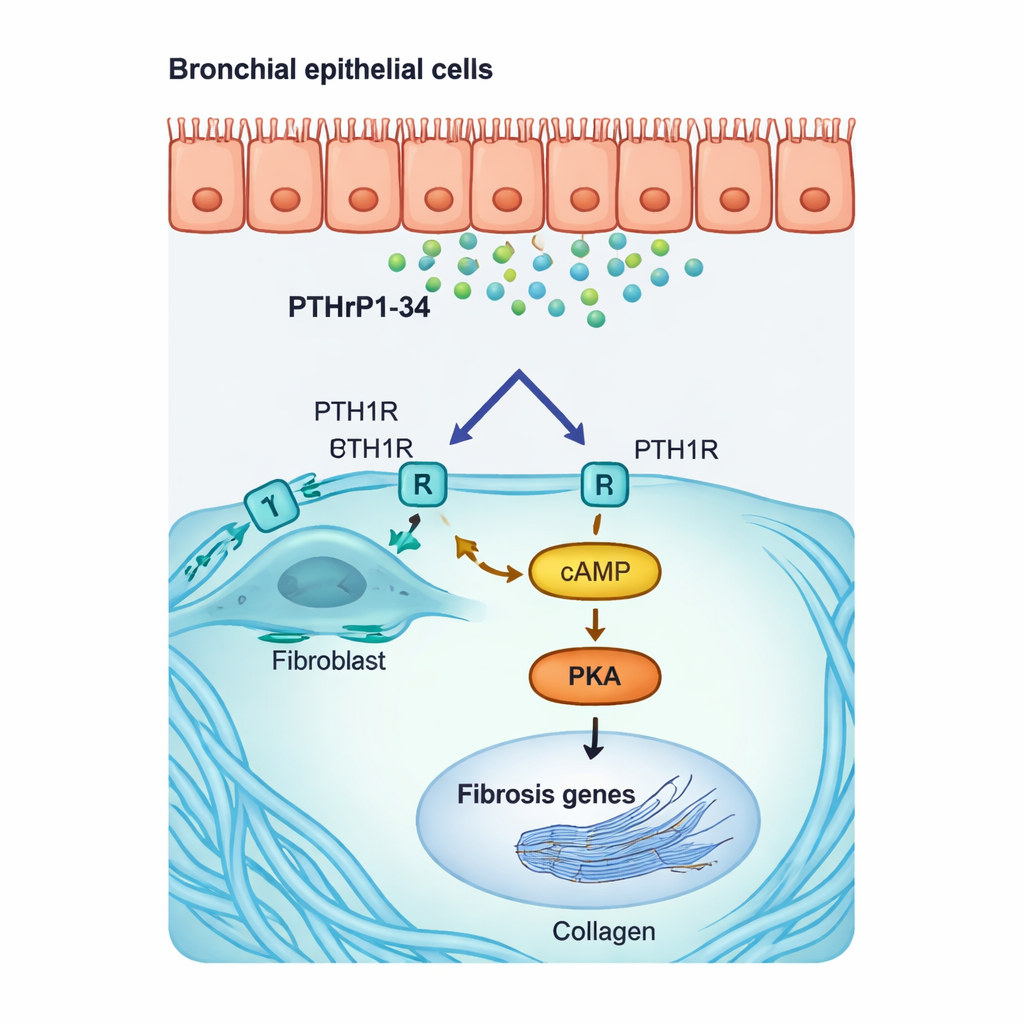
Beleg in erkrankten Lungen und neue Therapieansätze
Um zu prüfen, ob dieser Weg in ganzen Lungen relevant ist, nutzten die Forscher Mausmodelle von Lungenverletzung durch Bleomycin. Als die Lungen der Tiere von früher Entzündung zu dichter Vernarbung übergingen, stiegen die PTHrP1‑34‑Spiegel deutlich in den Atemwegen und im Lungengewebe, aber nicht im Blutkreislauf, was auf einen lokalen, lungenspezifischen Anstieg hindeutet. Die direkte Verabreichung von PTHrP1‑34 in die Atemwege trieb die Lunge in Richtung Fibrose, und in Kombination mit Bleomycin verschlechterte sich die Vernarbung und Kollagenablagerung deutlich. Das Team testete dann drei Wege, dieses schädliche Signal zu unterbrechen: einen neutralisierenden Antikörper, der PTHrP1‑34 bindet, ein kurzes Peptid, das dessen Rezeptor blockiert (PTHrP7‑34), und einen Gen-Silencing‑Ansatz, der die PTHrP‑Produktion in Atemwegszellen reduziert. In jedem Fall entwickelten die Mäuse weniger Lungenvernarbung, hatten geringere Kollagenwerte und behielten ein besseres Körpergewicht, mit Effekten, die mit denen von Nintedanib – einem zugelassenen IPF‑Medikament – vergleichbar waren oder diese sogar übertrafen.
Was das für die zukünftige Versorgung bedeutet
Für Nicht‑Spezialisten ist die Kernbotschaft: Das Bronchialepithel ist nicht nur ein passiver Luftleiter; es kann aktiv die Lungenvernarbung vorantreiben, indem es kraftvolle chemische Signale aussendet. Diese Studie identifiziert PTHrP1‑34 als eines dieser Signale und zeigt, dass das Unterbrechen seiner Kommunikation mit Fibroblasten Fibrose in Tiermodellen verlangsamen oder sogar umkehren kann. Zwar sind weitere Untersuchungen nötig, um Sicherheit und Wirksamkeit beim Menschen zu bestätigen, doch das Anvisieren des PTHrP1‑34/PTH1R‑Signalwegs könnte eine völlig neue Klasse von Therapien für IPF hinzufügen, die nicht nur das Fortschreiten verlangsamen, sondern einen seiner ursächlichen Treiber angehen.
Zitation: Fang, XQ., Lim, S., Lee, YM. et al. Parathyroid hormone–related protein is a therapeutic target in idiopathic pulmonary fibrosis. Sig Transduct Target Ther 11, 67 (2026). https://doi.org/10.1038/s41392-026-02578-8
Schlüsselwörter: idiopathische Lungenfibrose, Lungenfibrose, parathyroidhormon‑verwandtes Protein, Fibroblastenaktivierung, Bronchialepithel