Clear Sky Science · de
Exogenes Epstein-Barr-Virus-Nukleärantigen 1 induziert ADAR1-getriebene Tumorresistenz gegen Immuntherapie
Warum ein häufiger Virus für die Krebstherapie relevant ist
Viele Krebsarten werden inzwischen mit Immuntherapeutika behandelt, die das körpereigene Immunsystem freisetzen. Dennoch profitieren die meisten Patientinnen und Patienten nicht, weil ihre Tumoren lernen, sich vor Immunangriffen zu verbergen. Diese Studie zeigt, wie ein sehr verbreiteter Virus, das Epstein-Barr-Virus (EBV), Tumoren dabei hilft, Immunabwehr abzuschalten und gegen diese wirksamen Medikamente resistent zu werden — und wie ein neuer Typ von Designer-Molekül diese Abwehr möglicherweise wieder aktivieren kann.
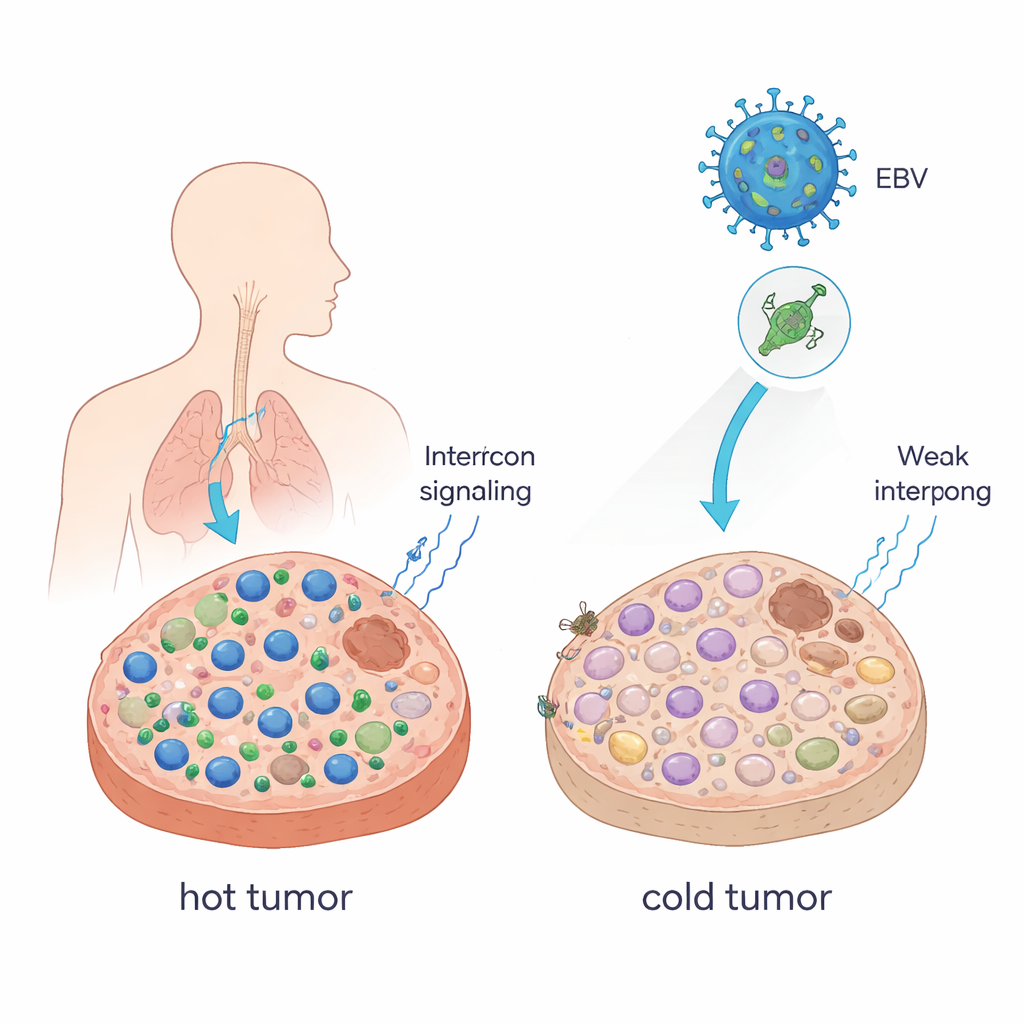
Aus heißen Tumoren werden kalte
Ärztinnen und Ärzte beschreiben Tumoren oft als „heiß“, wenn sie viele krebsabtötende T‑Zellen enthalten, und als „kalt“, wenn diese Zellen selten sind. Heiße Tumoren sprechen in der Regel gut auf Checkpoint‑Blockade‑Therapien (ICB) wie Anti‑PD‑1‑Antikörper an; kalte Tumoren dagegen oft nicht. Die Autorinnen und Autoren zeigten, dass ein einzelnes EBV‑Protein, EBNA1 genannt, Tumoren in diesen kälteren, ausweichenden Zustand drängen kann. Wenn sie Maus-Tumorzellen zwangen, EBNA1 zu produzieren, und diese in immunkompetenten Mäusen wuchsen, wurden die Tumoren größer, enthielten weniger CD8+-T‑Zellen und natürliche Killerzellen und wiesen mehr immunsuppressive Makrophagen auf. Interferone — entscheidende Botenstoffe zur Aktivierung von Immunzellen — waren ebenfalls stark reduziert. In Patientinnen‑ und Patientenproben des nasopharyngealen Karzinoms, einer Krebsform, die eng mit EBV verknüpft ist, zeigten EBNA1-exprimierende Tumoren ebenfalls weniger CD8+-T‑Zellen als normales Gewebe.
Eine virale Abkürzung in die RNA‑Kontrollmaschinerie der Zelle
Um zu verstehen, wie EBNA1 das Tumormilieu umgestaltet, suchten die Forschenden nach menschlichen Proteinen, die physisch mit ihm interagieren. Sie konzentrierten sich auf ein Protein namens IGF2BP3, das kleine chemische Markierungen (m6A) auf mRNAs liest und deren Stabilität oder Translation in Protein fördern kann. EBNA1 band in mehreren Zelltypen, einschließlich EBV-positiver Krebszellen, fest an IGF2BP3. Daten aus Tumoren von Patientinnen und Patienten zeigten, dass hohe IGF2BP3-Spiegel mit geringer Aktivität interferonbezogener Gene und niedrigerer CD8+-T‑Zell‑Infiltration einhergingen, was darauf hindeutet, dass diese Virus‑Wirt-Allianz die antitumorale Immunität dämpfen könnte.
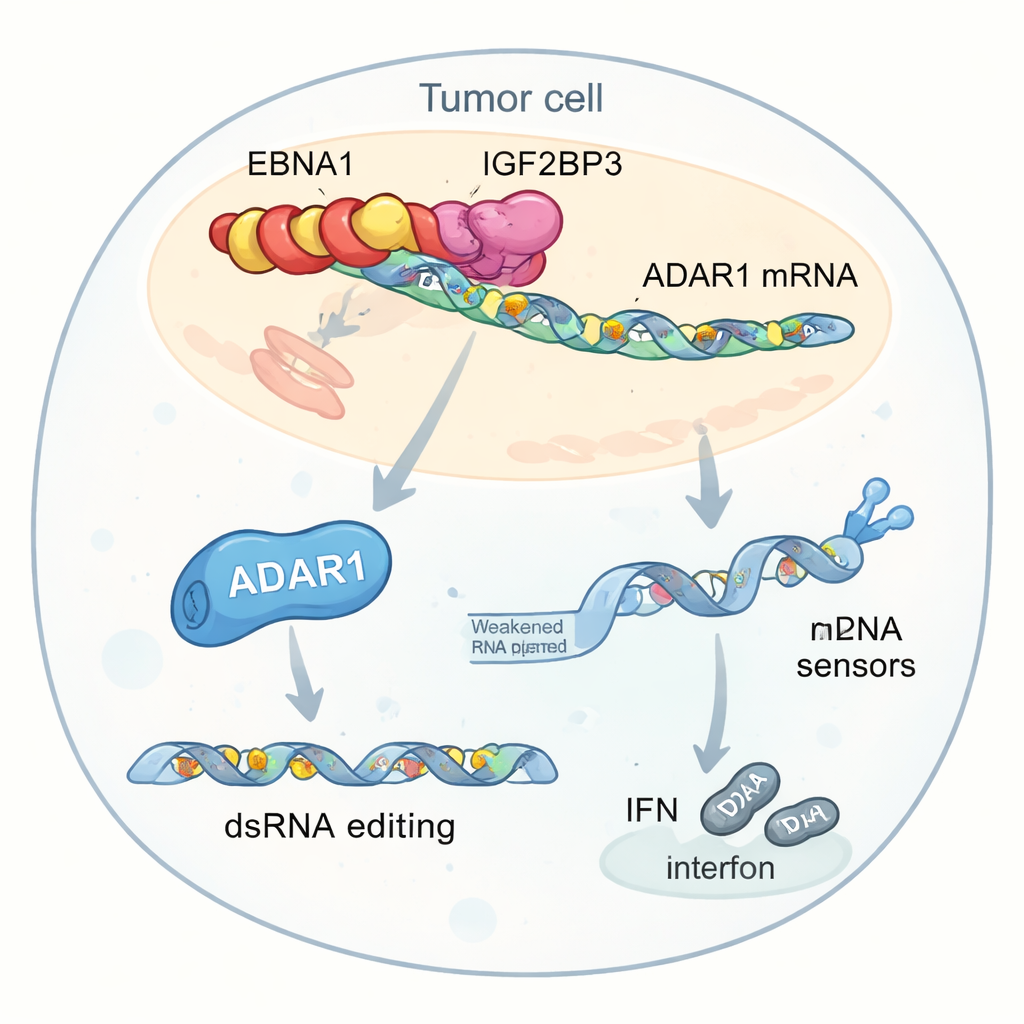
Steigerung eines RNA‑Editors, der Gefahrensignale stilllegt
Tiefere Analysen hoben ein wichtiges Ziel dieser Interaktion hervor: ADAR1, ein Enzym, das Doppelstrang‑RNA bearbeitet, indem es bestimmte „A“-Basen in „I“ umwandelt. Diese Editierung kann virenähnliche RNA in der Zelle weniger gefährlich erscheinen lassen und so Interferonantworten verringern. Die Autorinnen und Autoren fanden, dass EBNA1, IGF2BP3 und ein Translationsfaktor namens EIF4G1 einen Dreierkomplex auf der ADAR1‑mRNA bilden. Dieser Komplex erhöht die m6A‑Markierung, rekrutiert die Translationsmaschinerie und steigert selektiv die ADAR1‑Proteinproduktion, ohne deren RNA‑Spiegel anzuheben. Infolgedessen führen Tumorzellen mehr RNA‑Editierungen in repetitiven genetischen Elementen in der Nähe interferonbezogener Gene durch. Diese Editierungen reduzieren den Pool uneditierter Doppelstrang‑RNA, der normalerweise Sensoren wie MDA5 und PKR alarmieren würde, schwächen die Interferonproduktion und helfen Tumoren, sich vor Immunangriffen zu verbergen.
Weniger Interferon, schwächere Immuntherapie
Wenn EBNA1-exprimierende Tumorzellen im Labor T‑Zellen und Anti‑PD‑1‑Antikörpern ausgesetzt wurden, waren sie schwerer zu töten als Kontrollzellen und setzten weniger Interferon frei. Selbst bei direkter Behandlung mit Interferon reagierten EBNA1‑tragende Zellen weniger empfindlich, und ihre intrazellulären RNA‑Sensoren wurden weniger stark aktiviert. Eine Verringerung der ADAR1‑Mengen hob diese Effekte teilweise auf, stellte die Sensoraktivität und Interferonsignale wieder her. Genetische und Sequenzierungsexperimente bestätigten, dass EBNA1‑exprimierende Zellen mehr A‑zu‑I‑Editierungen in spezifischen RNA‑Regionen zeigten, insbesondere nach Interferon‑Stimulation, was die Vorstellung stützt, dass virale Hochregulierung von ADAR1 Gefahrensignale neutralisiert, die sonst starke Immunantworten auslösen würden.
Ein Designer‑Degrader, der den Immunangriff wieder aufweckt
Das Team fragte dann, ob das Entfernen von EBNA1 die Anfälligkeit der Tumoren für Immuntherapie wiederherstellen könnte. Sie entwickelten ein PROTAC‑Molekül, EP‑1215, das EBNA1 für den Abbau durch das zelluläre Entsorgungssystem markiert. In niedrigen Dosen degradierte EP‑1215 EBNA1 effizient und reduzierte ADAR1‑Proteinlevel. In Mausexperimenten hatte EP‑1215 allein nur begrenzte Wirkung auf EBNA1‑positive Tumoren, und Anti‑PD‑1 allein war ebenfalls schwach. In Kombination jedoch schrumpften die Tumoren deutlich, die CD8+-T‑Zell‑Infiltration nahm zu, und interferonproduzierende T‑Zellen wurden verstärkt. In humanisierten Mausmodellen mit menschlichen Immunzellen und EBV‑verknüpften Tumoren übertraf die Kombination erneut die Einzelsubstanzen, ohne offensichtliche Leber‑ oder Nierentoxizität.
Was das für die künftige Krebsbehandlung bedeutet
Für nicht‑Spezialisten ist die Botschaft: Ein verbreiteter Virus kann Krebszellen still und leise so umprogrammieren, dass interne Alarmsysteme gedämpft werden und chemische Signale, die sonst Immunzellen anziehen und aktivieren würden, abgeschaltet sind. EBNA1 erreicht dies, indem es einen Wirts‑RNA‑Reader (IGF2BP3) und einen Translationsfaktor (EIF4G1) kapert, um den RNA‑Editor ADAR1 zu überproduzieren, der genau jene RNA‑Strukturen editieren kann, für die Immun‑Sensoren ausgelegt sind. Durch den Abbau von EBNA1 mit einem maßgeschneiderten PROTAC wie EP‑1215 konnten die Autorinnen und Autoren diese Gefahrensignale wiederherstellen und resistente Tumoren erneut für vorhandene Checkpoint‑Medikamente empfindlich machen. Wenn ähnliche Strategien sich beim Menschen als sicher und wirksam erweisen, könnte das gezielte Ausschalten viraler Helfer wie EBNA1 einen neuen Weg eröffnen, kalte, EBV‑assoziierte Tumoren in heiße Ziele zu verwandeln, die moderne Immuntherapien erfolgreich angreifen können.
Zitation: Liu, C., Sun, Z., Li, C. et al. Exogenous Epstein–Barr virus nuclear antigen 1 induces ADAR1-driven tumor resistance against immunotherapy. Sig Transduct Target Ther 11, 63 (2026). https://doi.org/10.1038/s41392-026-02574-y
Schlüsselwörter: Epstein-Barr-Virus, Resistenz gegen Immuntherapie, ADAR1, RNA-Editierung, Nasopharynxkarzinom