Clear Sky Science · de
Häufige Genmutationen in 103 validierten kolorektalen Krebszelllinien
Warum diese Krebszelllinien wichtig sind
Das kolorektale Karzinom gehört zu den häufigsten Krebserkrankungen weltweit. Vieles von dem, was wir über sein Verhalten und seine Behandlung wissen, stammt aus Studien an im Labor gezüchteten Krebszelllinien. Diese Modelle können sich jedoch im Lauf der Zeit verändern. In dieser Studie wurden 103 weit verbreitete kolorektale Krebszelllinien sorgfältig überprüft und katalogisiert, wobei die wichtigsten Genmutationen, die das Tumorwachstum antreiben, kartiert wurden. Das Ergebnis ist ein detaillierter „Feldführer“, der Forschern hilft, das passende Modell für die jeweilige Fragestellung auszuwählen, mit dem Ziel, Laborbefunde wahrscheinlicher in künftigen Patientennutzen zu verwandeln.
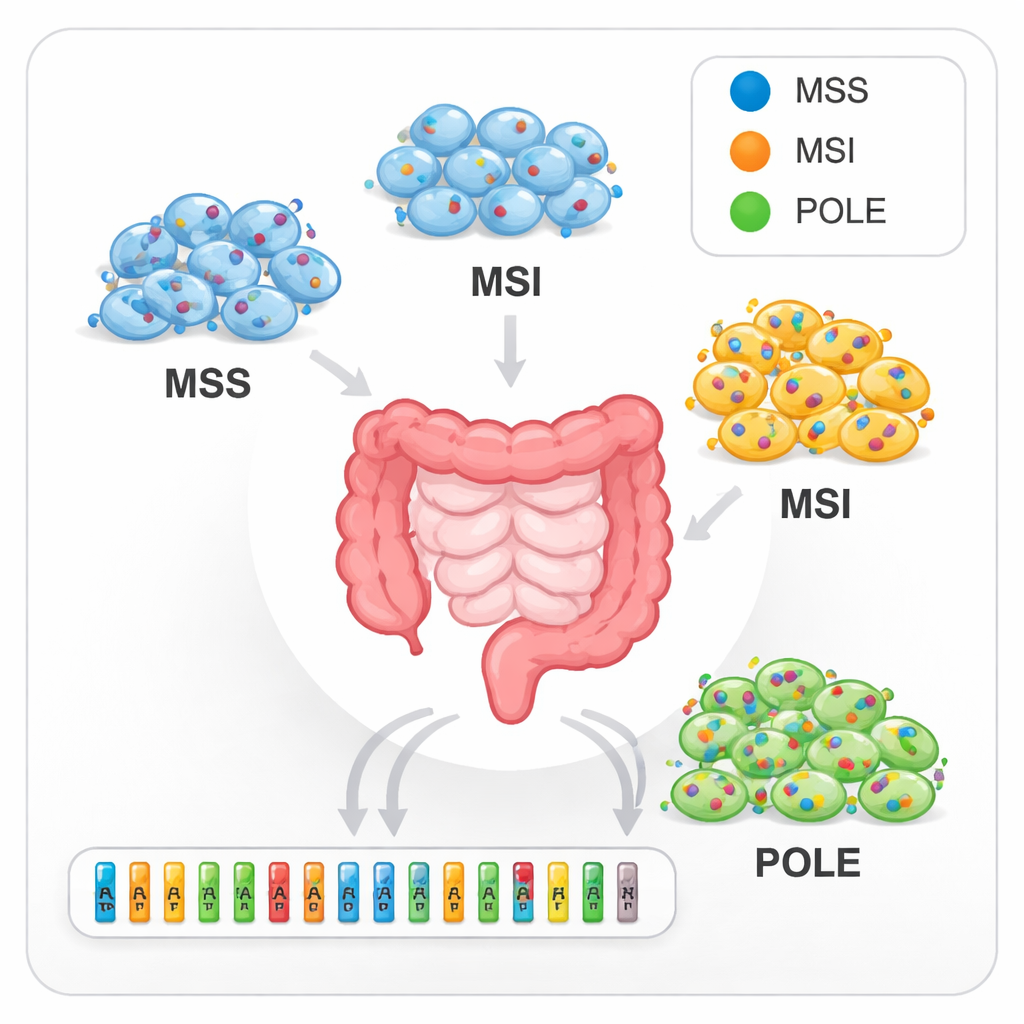
Ein Katalog von Krebsmodellen
Die Forschenden stellten 103 kolorektale Krebszelllinien zusammen, die gemeinsam die wichtigsten genetischen Subtypen widerspiegeln, die bei Patientinnen und Patienten vorkommen. Die meisten stammten aus primären Kolon- oder Rektumtumoren, die übrigen aus Metastasen. Zuerst verifizierte das Team, dass jede Zelllinie tatsächlich das war, was angegeben wurde, mithilfe von fingerabdruckähnlichen DNA-Markern, den sogenannten short tandem repeats. Anschließend nutzten sie tiefes, zielgerichtetes Sequenzieren und lasen ausgewählte DNA-Abschnitte im Mittel 575-fach, um 20 Gene zu untersuchen, die beim kolorektalen Karzinom bedeutsam sind. Diese hohe Tiefe ermöglichte es, selbst relativ seltene Mutationen innerhalb einer Zellpopulation zu entdecken und abzuschätzen, wie häufig jede Mutation unter den Zellen einer gegebenen Linie war.
Verschiedene Wege zur genetischen Unordnung
Kolorektale Karzinome können bei ihrer Entstehung unterschiedliche genetische Bahnen einschlagen. Etwa vier von fünf Tumoren zeigen chromosomale Instabilität mit großflächigen Genverlusten oder -gewinnen. Andere sind „hypermutiert“ und häufen viele kleine DNA-Veränderungen an. Diese Studie erfasste beide Muster. Sechzehn Zelllinien zeigten Microsatellite-Instabilität, eine Form der Hypermutation, die durch fehlerhafte DNA-Mismatch-Reparatur verursacht wird und zu häufigen kleinen Insertionen und Deletionen führt. Weitere fünf hatten Defekte im DNA-Proofreading-Gen POLE, was eine große Anzahl einzelner Basenveränderungen erzeugte. Die übrigen, nicht-hypermutierten Linien wiesen weniger Mutationen, dafür aber häufiger großskalige Veränderungen der DNA-Kopienzahl auf. Diese unterschiedlichen Signaturen ähnelten deutlich denen, die in Tumoren von Patientinnen und Patienten beobachtet werden.
Schlüssel-Treibergene und gefährliche Kombinationen
Über die 20 untersuchten Gene hinweg stimmten die Mutationsmuster mit großen klinischen Studien zum kolorektalen Karzinom überein. Tumorsuppressorgene wie APC, TP53 und SMAD4 waren häufig durch truncierende Mutationen betroffen, die das Protein zerstören, während klassische onkogene Treiber wie KRAS und BRAF häufiger einzelne Aminosäureänderungen trugen, die Signalwege an- oder abschalten. Nicht-hypermutierte Linien wiesen tendenziell einen höheren Anteil klar schädlicher, also pathogener Mutationen auf, die in den meisten oder allen Zellen der Kultur vorhanden waren. Im Gegensatz dazu trugen hypermutierte Linien viele zusätzliche „Passagier“-Veränderungen und mehr subklonale Mutationen, die nur in einem Teil der Zellen auftreten und die laufende Evolution in der Kultur widerspiegeln.
Verborgene Muster in der Geninteraktion
Durch die Analyse, welche Mutationen typischerweise zusammen auftreten oder einander ausschließen, konnten die Forschenden genetische Kombinationen identifizieren, die mit besonders aggressivem Krankheitsverlauf verknüpft sind. Beispielsweise kam die häufige Hotspot-Mutation BRAF p.V600 selten zusammen mit KRAS- oder NRAS-Mutationen vor, in einigen Linien trat sie jedoch zusammen mit truncierenden APC-Mutationen auf, was einem bei Patienten beobachteten Subtyp mit schlechter Prognose entspricht. Viele Zelllinien wiesen Dreifachtreffer in APC, TP53 und RAS-Genen auf, ein weiteres Merkmal hochriskanter Tumoren. Die Studie deckte außerdem ein charakteristisches „Double-Hit“-Muster in APC auf: zwei truncierende Mutationen waren so angeordnet, dass zumindest eine β-Catenin-Bindungsregion erhalten blieb, was zu einer „genau richtig“-Einstellung der WNT-Signalaktivität passt, die das Tumorwachstum begünstigt. Die Analyse der Kopienzahlen zeigte häufige Amplifikationen wachstumsbezogener Gene wie MYC und EGFR sowie den Verlust ganzer Kopien von Tumorsuppressorgenen, besonders in nicht-hypermutierten Linien.
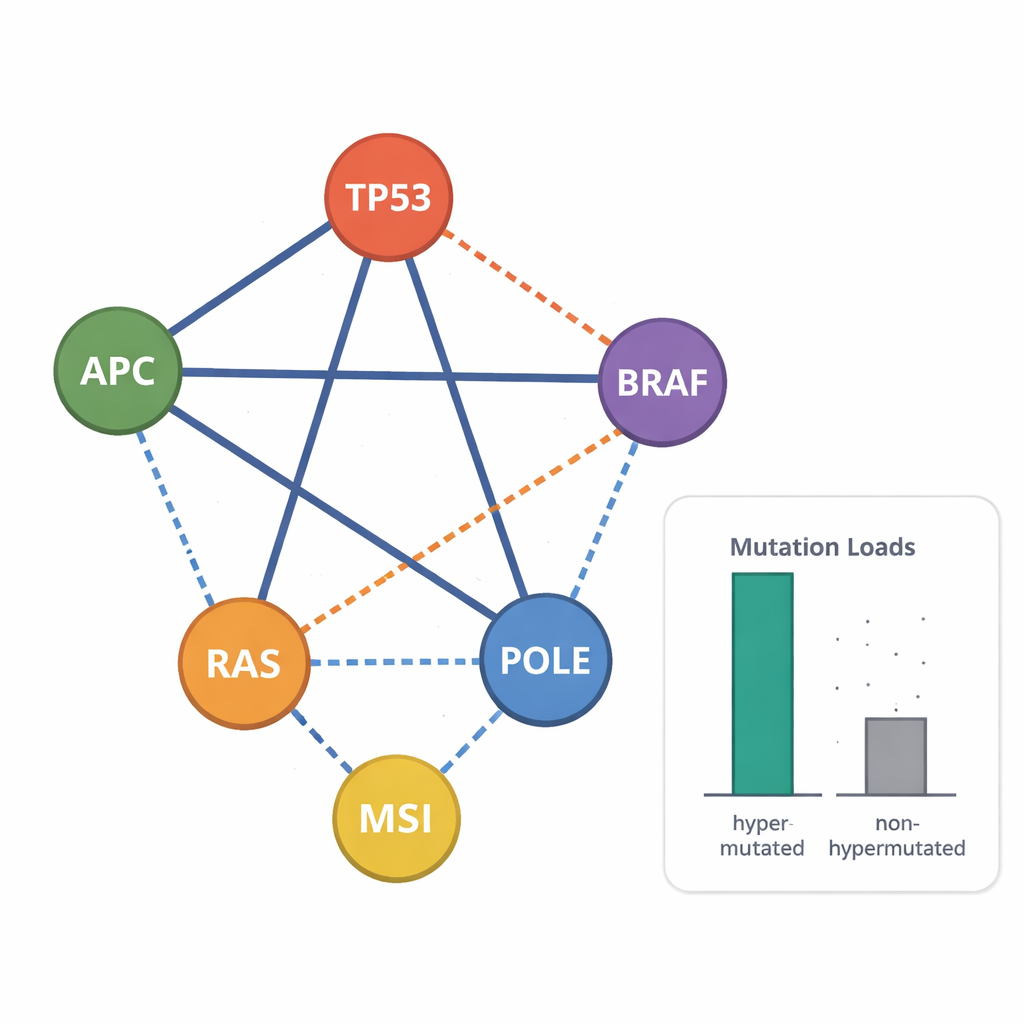
Was das für zukünftige Forschung bedeutet
Für Wissenschaftlerinnen und Wissenschaftler, die Experimente planen, lautet die Schlussfolgerung: Nicht alle kolorektalen Krebszelllinien sind gleich. Hypermutierte Modelle sind genetisch sehr heterogen und können viele niedrigfrequente Veränderungen enthalten, die die Wirkung einzelner Mutationen verschleiern. Nicht-hypermutierte Linien hingegen tragen tendenziell weniger, dafür stärkere und gleichmäßigere Treibermutationen. Indem diese Arbeit eine sorgfältig validierte Karte liefert, welche Gene verändert sind, auf welche Weise und wie häufig jede Mutation in jeder Kultur vorkommt, ermöglicht sie Forschenden, die am besten geeigneten Modelle für Wirkstofftests, biologische Untersuchungen und die Entwicklung neuer zielgerichteter oder immuntherapeutischer Ansätze beim kolorektalen Karzinom auszuwählen.
Zitation: Kranjec, C., Eilertsen, I.A., Nunes, L. et al. Common gene mutations in 103 authenticated colorectal cancer cell lines. Oncogenesis 15, 8 (2026). https://doi.org/10.1038/s41389-026-00599-0
Schlüsselwörter: kolorektales Karzinom, Krebszelllinien, Genmutationen, Tumorsubtypen, präzisionsonkologie