Clear Sky Science · de
PMM2 interagiert mit TRIM28, um E2F4 zu rekrutieren und KIFC3-vermittelte Tumor-Glykolyse sowie das Fortschreiten von kolorektalem Krebs zu fördern
Warum diese Krebs-Entdeckung wichtig ist
Kolorektales Karzinom gehört zu den tödlichsten Krebsarten weltweit, zum Teil weil viele Tumoren lernen, die Energiesysteme des Körpers zu kapern, um ungebremstes Wachstum zu ermöglichen. Diese Studie zeigt, wie ein wenig bekanntes Enzym, PMM2, kolorektalen Tumoren hilft, Zucker aggressiver zu verbrennen und zu streuen, und warum es damit ein vielversprechendes Ziel für zukünftige Medikamente und Diagnostika darstellt.
Eine zuckerhungrige Tumormaschine
Krebszellen verändern oft die Art und Weise, wie sie Glukose nutzen, und bevorzugen eine schnelle, energieineffiziente Form des Zuckerabbaus, die als Glykolyse bekannt ist. Die Forschenden begannen damit, tausende Gene in kolorektalen Tumorproben mit umliegendem gesundem Gewebe zu vergleichen. PMM2, ein Enzym, das normalerweise bei der Anheftung von Zuckerketten an Proteine mitwirkt, fiel als eines der am stärksten hochregulierten Gene im Tumor auf. Tumorzellen mit zusätzlichem PMM2 wuchsen schneller, bildeten mehr Kolonien und breiteten sich in Zellkultur häufiger aus, während Zellen, bei denen PMM2 abgeschaltet wurde, langsamer wuchsen, weniger migrierten und anfälliger für Zelltod waren.
Wie Tumorzellen die Zuckerverwertung beschleunigen
Als das Team die PMM2-Spiegel in kolorektalen Krebszellen verringerte, nahmen die Zellen weniger Glukose auf, erzeugten weniger ATP (ihre wichtigste Energiewährung) und schütteten weniger Laktat aus, das Abbauprodukt der Glykolyse. Empfindliche metabolische Messungen bestätigten, dass die allgemeine Ansäuerung des umgebenden Mediums abnahm, während die Sauerstoffnutzung stieg — ein Hinweis darauf, dass die Zellen sich von der beschleunigten Glykolyse hin zu normalerer Atmung verschoben. Wichtige Glykolyse-unterstützende Proteine, PKM2 und LDHA, fielen ebenfalls ab. Überraschenderweise konnte sogar eine katalytisch „tote“ Version von PMM2 dieses zuckerhungrige Verhalten fördern, was zeigt, dass die Rolle des Enzyms im Krebs nicht von seiner üblichen Chemie, sondern von seinen Bindungspartnern innerhalb der Zelle abhängt.
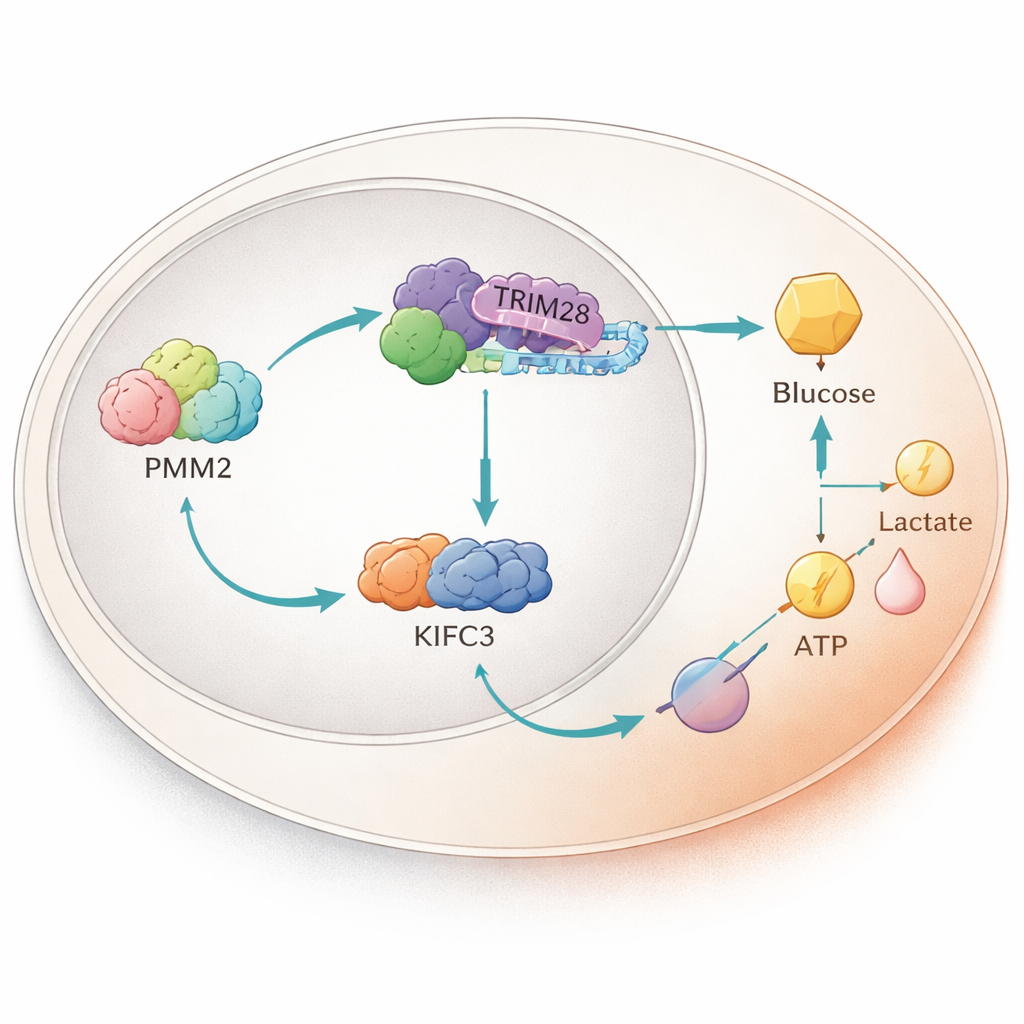
Eine Protein-Weiterleitung im Zellkern
Bei tiefergehenden Untersuchungen fanden die Wissenschaftler heraus, dass PMM2 physisch an ein anderes Protein namens TRIM28 bindet, das in den Zellkern wandern und die Genaktivität beeinflussen kann. PMM2 fördert die Anreicherung von TRIM28 im Kern, wo TRIM28 mit einem Transkriptionsfaktor, E2F4, zusammenarbeitet. Gemeinsam steigern diese drei die Produktion eines Motorproteins namens KIFC3, indem sie an einen spezifischen Abschnitt seiner DNA-Regionskontrolle binden. Experimente, die den PMM2-Bereich entfernten, der für die TRIM28-Bindung nötig ist, beseitigten PMM2s Fähigkeit, Glykolyse und Zellwachstum zu steigern, was unterstreicht, dass diese Proteinpartnerschaft — nicht PMM2s klassische Enzymfunktion — den Tumorvorteil antreibt.
Das Hochregeln eines zentralen Stoffwechsel-Schalters
KIFC3, sonst eher bekannt für seine Rolle beim Transport von Fracht entlang des zellulären Gerüsts, erwies sich als entscheidender metabolischer Schalter. Als die Forschenden KIFC3 reduzierten, konsumierten kolorektale Krebszellen weniger Glukose, produzierten weniger ATP und Laktat und zeigten geringere glykolytische Aktivität, während ihre Sauerstoffnutzung zunahm. Wichtig: Die Stilllegung von KIFC3 hob zum Teil die durch PMM2 bewirkte Steigerung von Glykolyse und Wachstums-Vorteil auf. In Mäusen, die mit menschlichen kolorektalen Krebszellen transplantiert wurden, wuchsen Tumore mit zusätzlichem PMM2 größer, doch dieser Effekt war abgeschwächt, wenn KIFC3 herunterreguliert wurde. Tumorproben dieser Tiere zeigten höhere Level von PMM2, KIFC3 und Glykolysemarkern und verbanden damit die gesamte Ereigniskette auch im lebenden Gewebe.
Von Labor-Modellen zu Patientenproben
Um die Arbeit näher an die Klinik zu bringen, erzeugte das Team miniaturisierte dreidimensionale Tumoren — sogenannte Organoide — aus Patientenproben kolorektaler Karzinome. Organoide mit höheren PMM2- und KIFC3-Spiegeln wuchsen schneller und produzierten mehr ATP und Laktat als solche mit niedrigeren Spiegeln. Eine erzwungene Überexpression von PMM2 erhöhte KIFC3 und Glykolyse, während die Reduktion von PMM2 die gegenteiligen Effekte zeigte. Analysen von Patienten-Tumorarrays zeigten außerdem, dass hohe PMM2-Werte mit fortgeschrittenerem Krankheitsstadium, Lymphknotenbefall und kürzerem Gesamtüberleben verbunden waren — ein Hinweis darauf, dass PMM2 ein starker Biomarker-Kandidat ist.
Was das für die künftige Behandlung bedeutet
Vereinfacht gesagt zeigt diese Studie, dass viele kolorektale Tumoren offenbar PMM2 in eine nukleare Protein-Weiterleitung — über TRIM28 und E2F4 — einschleusen, um KIFC3 hochzufahren und damit ihre zuckerverbrennende Maschinerie anzutreiben. Dieser metabolische Schub fördert Tumorwachstum und -ausbreitung. Weil dieser Weg von Proteininteraktionen und nicht von der üblichen Enzymfunktion von PMM2 abhängt, eröffnen sich neue therapeutische Ansätze: Kleine Moleküle, Peptide oder Degrader-Wirkstoffe, die die Bindung von PMM2 an TRIM28 stören, E2F4 den Zugang zur DNA blockieren oder die KIFC3-Aktivität dämpfen, könnten prinzipiell Tumoren ihrer bevorzugten Treibstoffquelle berauben. Solche Behandlungen sind bisher noch nicht verfügbar, doch die PMM2–TRIM28–E2F4–KIFC3-Kette steht nun als vielversprechende Landkarte für präzisere und stoffwechselorientierte Strategien gegen kolorektalen Krebs im Fokus.
Zitation: Peng, Z., Ma, B., Song, Z. et al. PMM2 interacts with TRIM28 to recruit E2F4 and promote KIFC3-mediated tumor glycolysis and colorectal cancer progression. Oncogene 45, 1145–1160 (2026). https://doi.org/10.1038/s41388-026-03707-x
Schlüsselwörter: kolorektales Karzinom, Tumorstoffwechsel, Glykolyse, onkogene Signalgebung, Biomarker