Clear Sky Science · de
CDK13 treibt das klarzellige Nierenkarzinom voran durch METTL16-vermittelte m6A-Modifikation der ACLY-mRNA
Warum fettgefüllte Nierentumoren wichtig sind
Klarzelliges Nierenkrebsgewebe erscheint unter dem Mikroskop oft hell und ölig, weil die Zellen mit Fett gefüllt sind. Dieses ungewöhnliche Erscheinungsbild ist nicht nur kosmetisch; es spiegelt eine tiefere Umprogrammierung des Kraftstoffmanagements dieser Krebszellen wider. Diese Studie stellt eine einfache, aber wichtige Frage: Welcher molekulare Schalter veranlasst Nierentumorzellen dazu, Fett anzusammeln, und lässt sich dieser Schalter ausschalten, um das Fortschreiten der Krankheit zu verlangsamen?
Ein verborgener Dirigent des Tumorwachstums
Die Forschenden konzentrierten sich auf ein Protein namens CDK13, das zur Familie von Enzymen gehört, die normalerweise die Zellteilung unterstützen. Durch die Analyse großer Patientendatensätze und Tumorproben fanden sie heraus, dass die CDK13-Spiegel in klarzelligen Nierenkarzinomen konstant höher sind als im normalen Nierengewebe. Patienten mit Tumoren, die mehr CDK13 aufwiesen, hatten tendenziell größere, fortgeschrittenere Tumoren und schlechtere Prognosen. Wenn das Team CDK13 in Nierenkrebszelllinien reduzierte, wuchsen die Zellen langsamer und hatten Schwierigkeiten, den Zellzyklus zu durchlaufen, was darauf hindeutet, dass CDK13 als verborgener Dirigent sowohl Wachstum als auch Überleben koordiniert.
Von Zucker zu Fett: Umprogrammierung der zellulären Kraftfabrik
Da klarzellige Nierentumoren reich an Lipiden sind, untersuchte das Team, ob CDK13 auch die Fettsynthese dieser Zellen steuert. Mithilfe einer Kombination aus Genexpressionsprofilen und mikroskopischen Fettfärbungen zeigten sie, dass eine Erhöhung von CDK13 die Ansammlung von Lipidtröpfchen in Krebszellen fördert, während die Reduktion von CDK13 den gegenteiligen Effekt hat. CDK13 beeinflusste stark ein Enzym namens ACLY, das ein häufiges Stoffwechselzwischenprodukt in Acetyl-CoA umwandelt — den Ausgangsstoff für die Synthese von Fettsäuren und Cholesterin. Hohe CDK13-Spiegel gingen einher mit hohen ACLY-Spiegeln in Patientenproben, und beide Proteine konzentrierten sich in denselben Tumorgeweberegionen. Wenn ACLY künstlich hochreguliert wurde, kompensierte es viele der Wachstums- und Fettspeicherdefekte, die durch CDK13-Verlust entstanden waren, womit ACLY als zentraler nachgeschalteter Effektormechanismus dieses Weges positioniert wird. 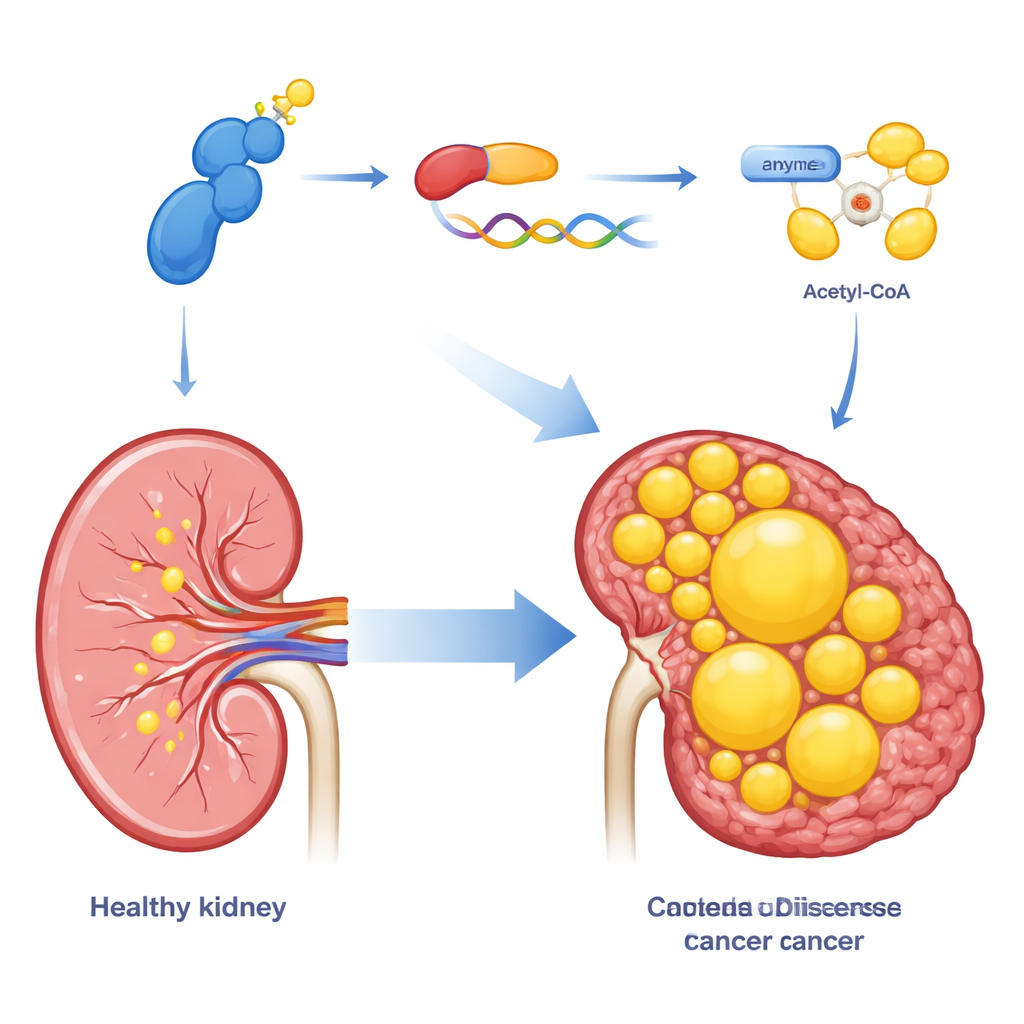
Ein geschichtetes Nachrichtensystem innerhalb der Krebszellen
Anstatt direkt als einfacher Ein-/Ausschalter auf ACLY zu wirken, entfaltet CDK13 seine Wirkung über ein geschichtetes Nachrichtensystem auf Basis von RNA, dem Molekül, das genetische Anweisungen von der DNA zu den Proteinfabriken trägt. Die Autorinnen und Autoren entdeckten, dass CDK13 physisch an ein weiteres Enzym, METTL16, bindet und es chemisch modifiziert; METTL16 versieht spezifische RNA-Nachrichten mit kleinen chemischen Markierungen, sogenannten Methylgruppen. CDK13 fügt METTL16 an einer bestimmten Stelle eine Phosphatmarke hinzu, wodurch METTL16 aktiver wird. Im Gegenzug setzt METTL16 zusätzliche Methylierungen auf die RNA-Bauanleitung von ACLY. Diese Markierungen verändern nicht den genetischen Code selbst, wohl aber, wie die Zelle diese Nachricht behandelt. Ein drittes Protein, YTHDC2, erkennt die markierte ACLY-RNA und schützt sie vor dem Abbau, sodass über die Zeit mehr ACLY-Protein produziert werden kann. Diese Kaskade — CDK13 aktiviert METTL16, METTL16 markiert die ACLY-RNA und YTHDC2 schützt diese markierte Nachricht — erzeugt eine starke Rückkopplung zur Förderung der Fettsynthese. 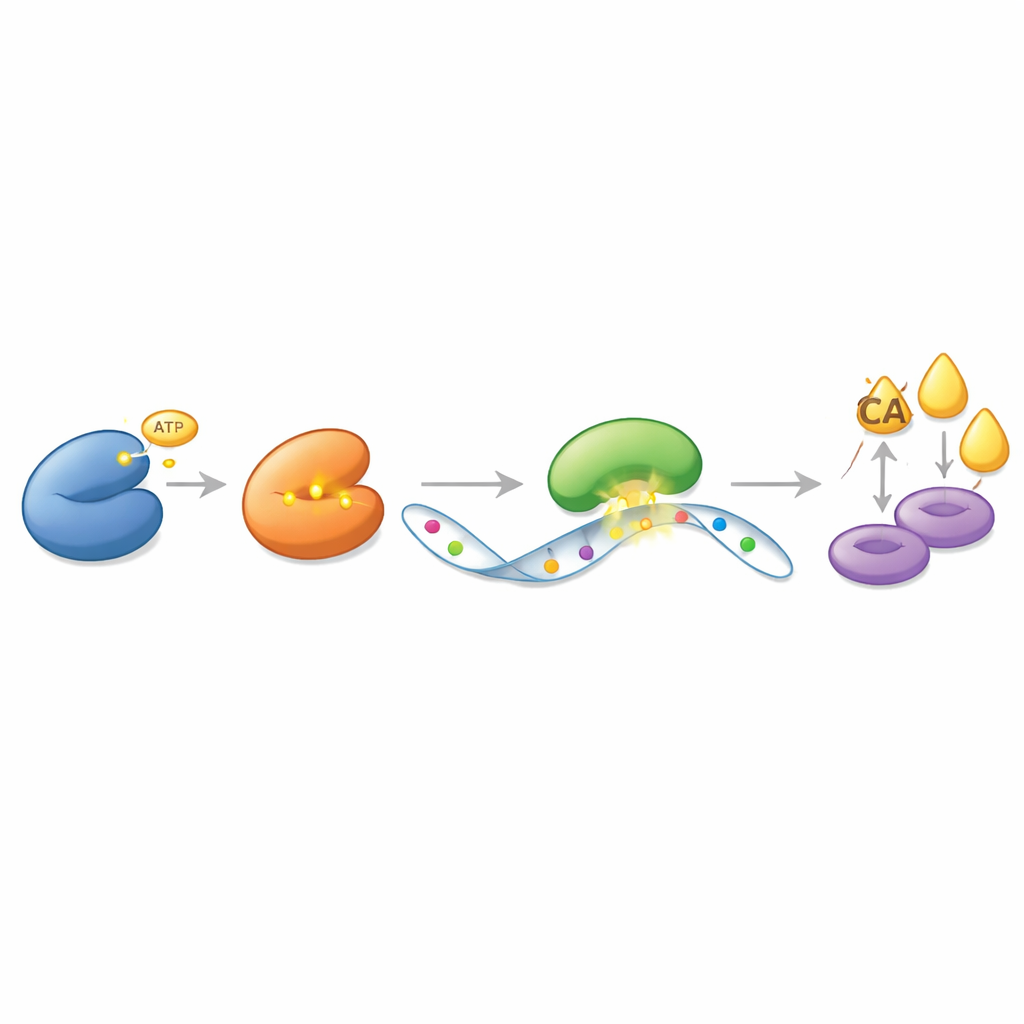
Prüfung der Kette in Zellen, Mäusen und Patientenproben
Die Stärke der Arbeit liegt darin, wie gründlich die Autorinnen und Autoren diese Ereigniskette überprüften. In kultivierten Nierenkrebszellen reduzierte die Störung irgendeines Teils der CDK13–METTL16–ACLY-Achse die Lipidtröpfchen und verlangsamte die Proliferation. In Mäusen, die mit menschlichen Nierenkrebszellen transplantiert wurden, schrumpften Tumoren und deren Fettdepots, wenn CDK13 oder ACLY allein blockiert wurden; die gleichzeitige Blockade beider führte zu einem noch stärkeren Effekt. Das Team verwendete außerdem eine kleinmolekulare Verbindung, 1NM-PP1, die die CDK13-Aktivität hemmt. Dieser wirkstoffähnliche Stoff verringerte die aktivierende Markierung auf METTL16, senkte die ACLY-Spiegel und unterdrückte das Tumorwachstum, besonders in Kombination mit METTL16-Reduktion. In Patienten-Datensätzen stiegen und fielen CDK13, METTL16 und ACLY häufig gemeinsam, was die Idee untermauert, dass diese Achse in echten Tumoren aktiv ist und nicht nur in Labor‑Modellen.
Was das für zukünftige Behandlungen bedeuten könnte
Für Nicht‑Spezialisten ist die Kernbotschaft, dass diese Studie einen neuen Kontrollknopf für die „Fettfabrik“ in klarzelligen Nierentumoren offenlegt. Statt nur die Enzyme anzugreifen, die Lipide herstellen, zeigen die Forschenden eine übergeordnete Befehlskette, die die Stabilität der Anweisungen für diese Enzyme sichert. Durch das Unterbrechen der CDK13–METTL16–ACLY‑Achse könnte man Tumoren die für ihr Wachstum und die Ausbreitung notwendigen Fette entziehen, während normale Zellen weniger betroffen bleiben. Obwohl die Arbeit noch präklinisch ist und 1NM-PP1 noch kein zugelassenes Nierenkrebsmedikament ist, weisen die Ergebnisse auf neue Strategien hin, die Kinasehemmer mit Wirkstoffen kombinieren, die RNA-modifizierende Enzyme anvisieren, und damit einen präziseren Ansatz für diese metabolisch getriebene Form des Nierenkrebses bieten.
Zitation: Chen, J., Liu, H., Zhang, Y. et al. CDK13 drives clear cell renal carcinoma through METTL16-mediated m6A modification of ACLY mRNA. Exp Mol Med 58, 472–486 (2026). https://doi.org/10.1038/s12276-025-01634-7
Schlüsselwörter: klarzelliges Nierenzellkarzinom, Lipidstoffwechsel, CDK13, RNA-Methylierung, ACLY