Clear Sky Science · ar
تباين المعدلات وأخطاء التسلسل المتكررة في علم النشوء والتطور على مقياس الجائحة
لماذا هذا مهم في التفشي المستقبلي
عندما ينتشر فيروس جديد في جميع أنحاء العالم، يتسابق العلماء لقراءة شيفرته الجينية وإعادة بناء شجرة أصله. تساعد تلك الأشجار في تتبع كيفية ظهور المتغيرات، ومدى سرعتها في الانتشار، وما إذا كانت إجراءات السيطرة فعالة. لكن أثناء جائحة كوفيد‑19، قامت المختبرات بتسلسل ملايين جينومات SARS‑CoV‑2 بسرعة كبيرة إلى حد أن أخطاء وخاصيات مخفية في البيانات بدأت تشوّه الصورة. تُقدّم هذه الورقة طرقًا جديدة لتنقية وتفسير مثل هذه المجموعات الجينية الضخمة، موفرة رؤية أوضح لكيفية تطور فيروس الجائحة وانتشاره بين السكان.
تحدي فهم ملايين الجينومات
يحوّل علم الأوبئة الجينومية جينومات الفيروس إلى معلومات عملية لاتخاذ القرارات الصحية العامة. بالنسبة لـ SARS‑CoV‑2، تم مشاركة أكثر من 20 مليون جينوم حول العالم. بُنيت أدوات التطور التقليدية لمشكلات أكثر اعتدالًا —مثل مقارنة الجينات بين الأنواع— وليست مهيأة للتعامل مع ملايين من التسلسلات الفيروسية المتقاربة التي تصل في الوقت الحقيقي. على هذا النطاق، تصبح مسألتان مزعجتين بشكل خاص. أولًا، بعض المواقع في الجينوم الفيروسي تتعرض لطفرات بمعدل أعلى بكثير من غيرها، مما قد يجعل فيروسات غير مرتبطة تبدو متشابهة بشكل غريب. ثانيًا، يمكن أن تُحاكي الأخطاء التقنية المتكررة في التسلسل ومعالجة البيانات طفرات حقيقية. كلا التأثيرين يولّدان «صدى زائفًا» في شجرة التطور، ما يخلق عدم يقين بشأن الفروع والتجمعات التي ينبغي الوثوق بها.
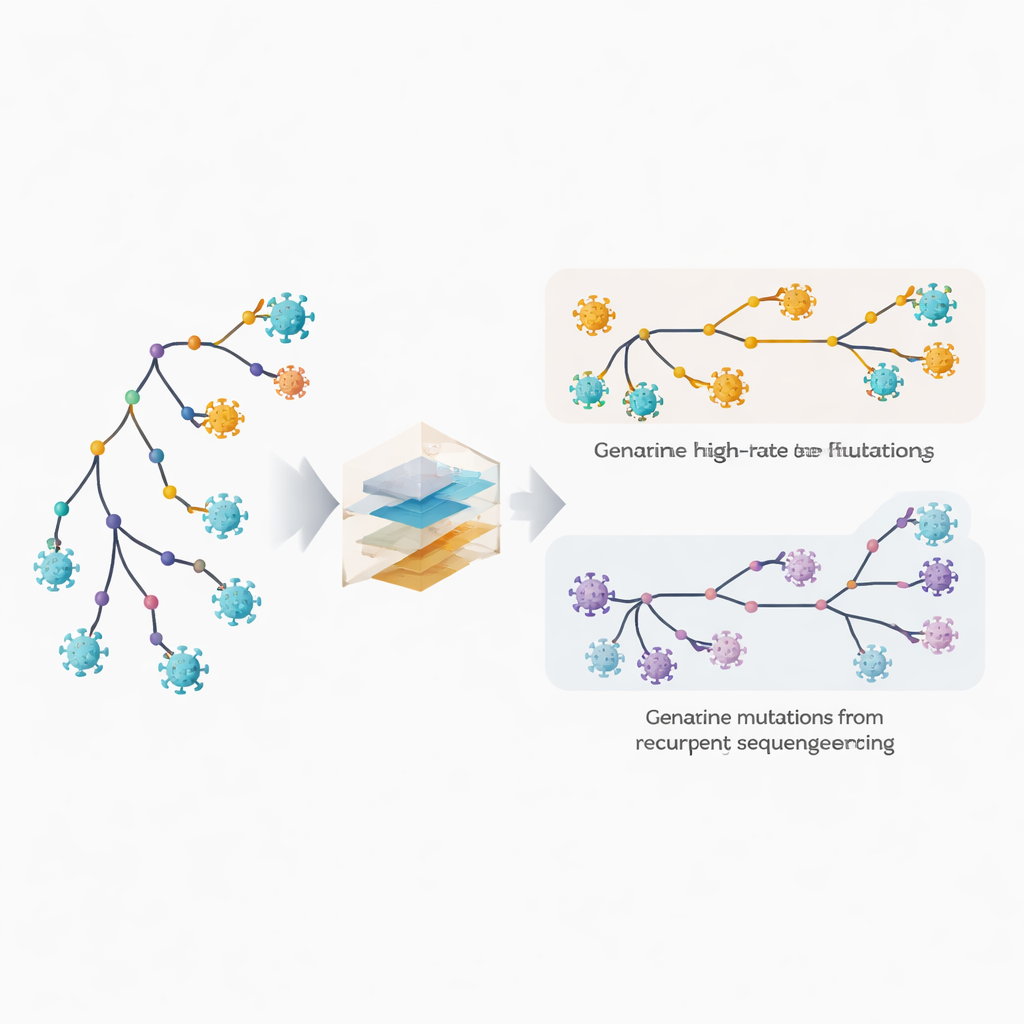
كشف المواقع سريعة التغير والأخطاء الخفية
وسع المؤلفون برنامجهم الفيلوجيني MAPLE بنماذج تعامل كل موضع في الجينوم الفيروسي على أنه يملك سلوكًا خاصًا به. بدلًا من افتراض عدد قليل من معدلات الطفرات المتوسطة، يقدّر الأسلوب معدلًا منفصلًا لكل موقع، مستفيدًا من الكم الهائل من الجينومات المتاحة. في الوقت نفسه، يسمح لكل موقع بأن يكون له احتمال خاص لحمل خطأ متكرر في التسلسل أو في استدعاء السلسلة التوافقية (consensus). الخدعة الأساسية هي مقارنة عدد المرات التي يظهر فيها تغيير على فروع داخلية عميقة في الشجرة، التي تعكس أحداثًا أقدم ومشتركة، مقابل تلك التي تظهر على الأطراف الخارجية الأبعد، والتي تمثل جينومات فردية. تميل الطفرات البيولوجية الحقيقية إلى التوزع بين الفروع الداخلية والطرفية، بينما تظهر الأخطاء التقنية في الغالب عند الأطراف. باستغلال هذا النمط، يمكن للطريقة فَصْل التطور السريع الحقيقي عن الأخطاء المتكررة.
خوارزميات أسرع لشجرة حياة مكتظة
كان التعامل مع ملايين الجينومات سيتطلب عادة قدرة حوسبة هائلة. للحفاظ على العملية عملية، أعاد الفريق تصميم طريقة تخزين MAPLE وتحديث معلومات التسلسل على الشجرة. بدلًا من مقارنة كل جينوم إلى مرجع ثابت واحد، يختار البرنامج «مراجع محلية» داخل الشجرة ويسجل الجينومات القريبة كاختلافات نسبية لتلك النقاط الثابتة. هذا التمثيل المدمج يسرّع المقارنات بين أجزاء بعيدة من الشجرة. تحسينات إضافية تصقل كيفية إضافة عينات جديدة إلى شجرة قائمة، وكيفية ضبط أطوال الفروع، وكيفية استكشاف أشكال شجرية بديلة محتملة، مع خيارات لتشغيل أقسى الخطوات المتطلبة بالتوازي عبر عدة أنوية معالجة.
اختبار الطريقة وتنقية بيانات العالم الحقيقي
للتحقق من فعالية نماذجهم، أنشأ المؤلفون أولًا مجموعات بيانات SARS‑CoV‑2 محاكاة واقعية بنماذج طفرة معروفة وأخطاء تسلسل مضمنة. في هذه الاختبارات، استعاد النهج الجديد أشجارًا تطورية أكثر صدقًا وحدد الأخطاء الفردية بدقة عالية، خصوصًا عند إدراج عشرات الآلاف من الجينومات أو أكثر. ثم انتقلوا إلى بيانات حقيقية، محللين ملايين تسلسلات SARS‑CoV‑2 التي كانت القراءات الأولية الخام متاحة لها. بمقارنة خطيْ بناء توافق مختلفيْن، حدّدوا مواضع جينومية محددة تتأثر مرارًا بتحفّرات مثل مشاكل ارتباط البرايمر أو استدعاء متحيّز نحو المرجع. تم قناع هذه المواقع المشبوهة من التحليل اللاحق، وتمت تصفية الجينومات التي أظهرت علامات تلوث أو عدوى مختلطة، مما أدى إلى محاذاة مَنقّحة تضم أكثر من مليوني تسلسل عالي الجودة.
صورة عالمية أوضح لشجرة عائلة الفيروس
باستخدام مجموعة البيانات المنقّحة، أعاد المؤلفون بناء شجرة فيلوجينية عالمية لـ SARS‑CoV‑2 ورسموا كيف ترتبط المتغيرات الرئيسية ببعضها. تقترح شجرتهم في بعض الأحيان علاقات تختلف بدقة عن الأشجار العامة السابقة، وغالبًا بطرق تتطلب عددًا أقل من أحداث الطفرة وتطابق النموذج الإحصائي بشكل أفضل. كما يبرز الإطار أماكن قد تكون فيها تسميات السلالات غير متوافقة مع التاريخ الجيني الأساسي، معلنًا عن احتمالات وجود متعادلات (recombinants) أو جينومات إشكالية تحتاج لفحص أدق. وعلى الرغم من بقاء بعض التحديات —مثل الإفراط في الضبط عندما تكون البيانات شحيحة أو تأثير العينات الملوثة بشدة— تظهر الدراسة أنه صار ممكنًا الآن بناء أشجار تطورية أكثر موثوقية على مستوى الجائحة. للخلاصة للقارئ العام: التعامل الأفضل مع الأخطاء ونقاط الطفرات الساخنة يؤدي إلى فهم أدق لكيفية انتشار وتغير العوامل الممرضة، مما يساعد العلماء والوكالات الصحية على الاستجابة بسرعة وثقة أكبر في التفشيات المستقبلية.
الاستشهاد: De Maio, N., Willemsen, M., Martin, S. et al. Rate variation and recurrent sequence errors in pandemic-scale phylogenetics. Nat Methods 23, 565–573 (2026). https://doi.org/10.1038/s41592-025-02932-8
الكلمات المفتاحية: علم جينات SARS‑CoV‑2, طرق علم النشوء والتطور, أخطاء التسلسل, تباين معدل الطفرات, علم الأوبئة الجينومية