Clear Sky Science · ar
DiNovo تمكّن من تسلسل الببتيدات الجديد بتغطية وثقة عاليتين عبر بروتيازات مرآوية وتعلّم عميق
رؤية البروتينات بتفصيل جديد
البروتينات هي الآلات الدقيقة التي تُبقي خلايانا على قيد الحياة، لكن قراءة مكوّناتها الأساسية بشكل كامل لا تزال أمراً أصعب مما قد يظن المرء. تقدّم هذه الورقة DiNovo، نظام برمجي جديد يساعد العلماء على «قراءة» شظايا البروتين بدقّة وموثوقية أكبر بكثير من ذي قبل. عبر الجمع بين خدعة بيوكيميائية ذكية والذكاء الاصطناعي الحديث، يعد الاكتشاف المحتمل لبروتينات مخفية، علامات مرضية، وحتى أهداف مناعية قد تغفلها الطرق التقليدية.
لماذا قراءة شظايا البروتين صعبة للغاية
تعتمد معظم تحليلات البروتين اليوم على تقطيع البروتينات إلى قطع أصغر تُسمى ببتيدات، ثم وزن شظاياها في جهاز الطيف الكتلي. من هذه الأوزان، تحاول الحواسيب إعادة بناء تسلسل الببتيد الأصلي، كحلّ صفقة كلمات متقاطعة من أدلة جزئية. الأساليب الحالية تفترض عادة أن الببتيدات مأخوذة من قواعد بيانات بروتينات معروفة، وهذا يعمل جيداً للبروتينات المألوفة لكنه يتعثر أمام البروتينات الجديدة أو غير المتوقعة. يُجنب ما يُسمى بالتسلسل من الصفر هذا القيد بمحاولة قراءة الببتيدات مباشرة من البيانات، لكنه كثيراً ما يقصر لأن بعض الشظايا مفقودة وبعض الببتيدات لا تُقطع نظيفاً أصلاً.
استخدام إنزيمات مرآوية لملء الفجوات
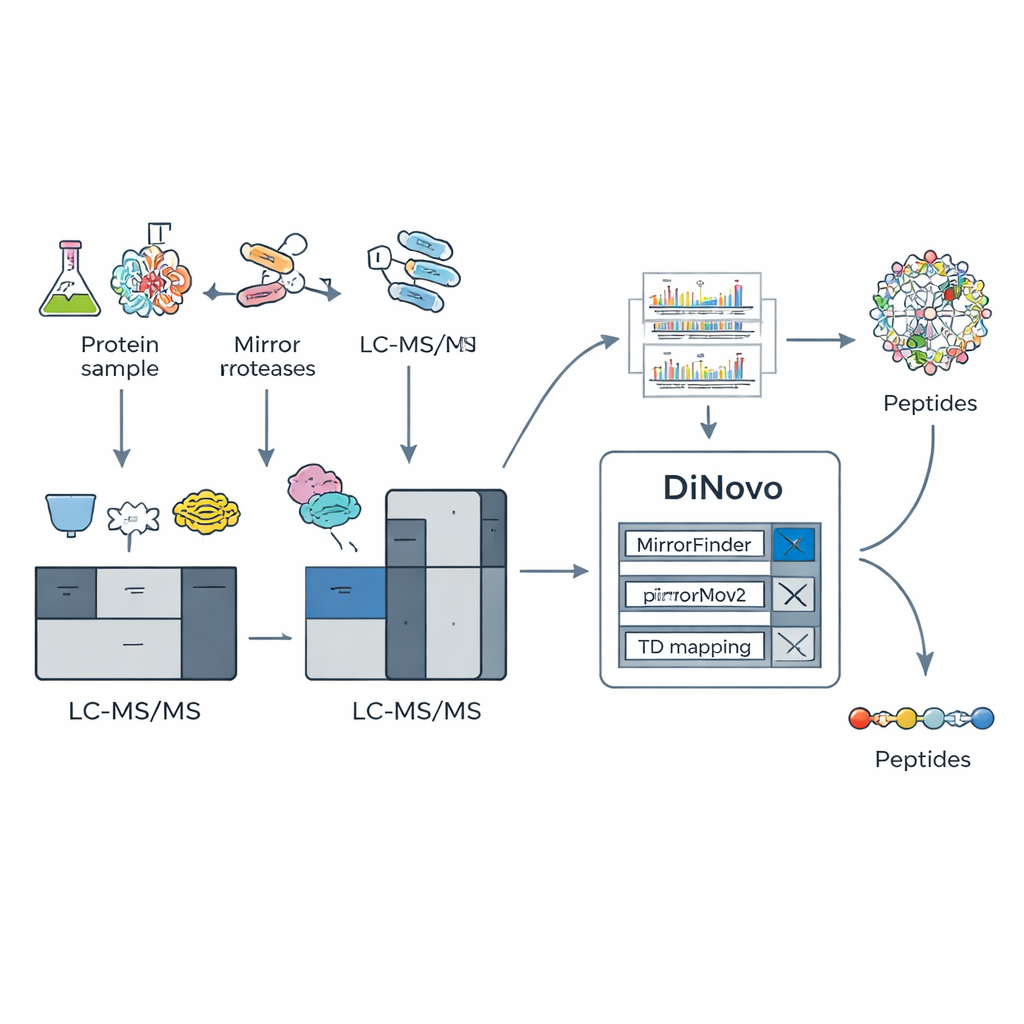
الفكرة الأساسية في DiNovo هي استخدام أزواج من «البروتيازات المرآوية» — أزواج من إنزيمات القطع التي تقطع البروتينات من جوانب متعاكسة لنفس نوع الحمض الأميني. على سبيل المثال، يقطع إنزيم واحد قبل الليسين مباشرة، بينما شريكه يقطع بعد الليسين. هذا يولّد ببتيدين مرتبطين لهما نفس الجزء الداخلي لكن بأطراف مختلفة. عند تحليل طيف الكتلة لهذين الببتيدين «المرآوين»، تحتوي الأطياف على أنماط شظايا مكملة: ما يكون مفقوداً في طيف واحد غالباً ما يظهر في الآخر. يبيّن المؤلفون أن الجمع بين مثل هذه الأزواج يمكن أن يدفع تغطية الشظايا لتقارب الاكتمال، مع دعم نحو 98% من القطوع الممكنة بواسطة إشارات تجريبية حقيقية، وهو أعلى بكثير من ما يُرى عند استخدام إنزيم واحد فقط.
خط أنابيب برمجي ذكي مخصص لبيانات المرآة
لاستغلال هذه الحيلة البيوكيميائية، بنى الفريق DiNovo كمسار عمل برمجي متكامل. أولاً، تُهضَم بروتينات من بكتيريا وخمائر باستخدام زوجين مرآويين من الإنزيمات، وتُحلّل الببتيدات الناتجة بواسطة طيف كتلي عالي الدقة. ثم يستخدم DiNovo وحدة تُسمى MirrorFinder للتعرّف تلقائياً على أي أزواج من الأطياف تأتي من ببتيدات مرآوية، معتمداً في ذلك على أنماط الإشارة بدلاً من أي تخمينات تسلسلية سابقة. بعد ذلك، يستخدم محركه الرئيسي للتسلسل من الصفر، MirrorNovo، التعلّم العميق لتفسير تلك الأطياف المزدوجة، بينما يوفر محرك احتياطي قائم على الرسوم البيانية، pNovoM2، خياراً أسرع يعمل على وحدة المعالجة المركزية فقط. معاً، تترجم هذه الأدوات القمم إلى تسلسلات أحماض أمينية وتفحص أيضاً الأطياف الفردية التي لم تُشكّل أزواجا واضحة، مستخرجة أكبر قدر ممكن من المعلومات.
قياس الثقة دون الاعتماد على قواعد بيانات قديمة
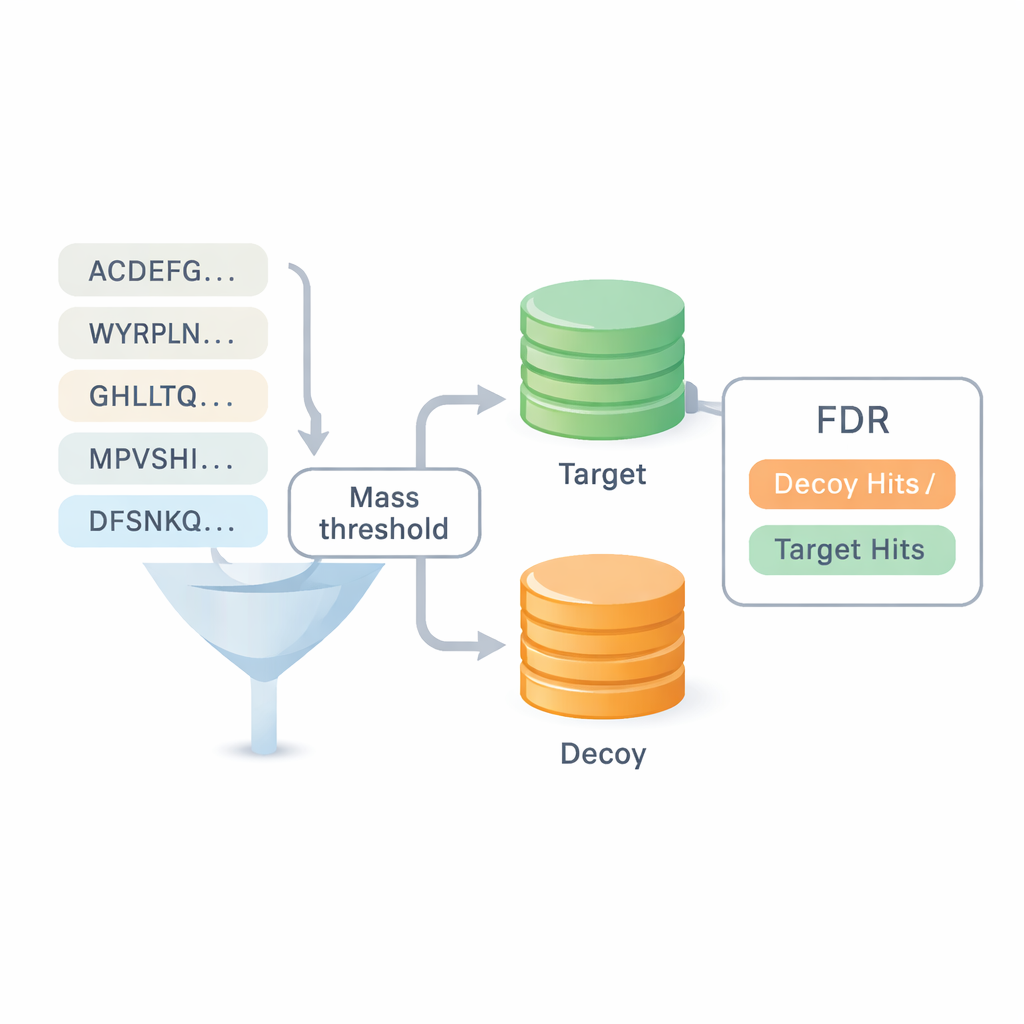
أحد أكبر الأسئلة في التسلسل من الصفر هو مدى موثوقية النتائج. تعتمد معظم المعايير الحالية على إعادة استخدام إجابات من بحث القواعد البيانية، ما يطمس الفاصل بين النهجين وقد يخفي أخطاء. يقدم DiNovo طريقة تحقق جودة مختلفة تسمى مطابقة الهدف-الطُعْن. هنا تُطابق الببتيدات المقروءة حديثاً إلى مجموعة مدمجة من تسلسلات بروتين حقيقية (هدف) ومُزيّفة ومنزوعة المعنى (طُعْن). بمقارنة كم مرة تستقر الببتيدات في المجموعة الحقيقية مقابل المجموعة المزيّفة، يمكن للبرنامج تقدير معدل الخطأ، أو معدل الاكتشاف الكاذب، دون الاعتماد على تعريفات سابقة. هذا يجعل من الممكن مقارنة DiNovo مباشرة مع برامج بحث قواعد البيانات القياسية تحت نفس ضوابط الخطأ.
ما يقدّمه DiNovo عملياً
في اختبارات على عينات بكتيرية وخميرة وأجسام مضادة، قرأ DiNovo باستمرار عددًا أكبر من الببتيدات والأحماض الأمينية مقارنة مع أدوات التسلسل من الصفر المعروفة التي تستخدم إنزيمًا واحدًا فقط. باستخدام زوجين مرآويين، أنتج ضعف إلى ثلاثة أضعاف عدد الأحماض الأمينية عالية الثقة مقارنة بإعداد كلاسيكي يعتمد فقط على التربسين، وحدد مزيداً من البروتينات عند مستويات خطأ مماثلة. وعند مقارنته مباشرة بثلاثة محركات بحث قواعد بيانات رائدة، وجد DiNovo أعداداً مماثلة من الأحماض الأمينية والبروتينات، وتوافقت معظم تتابعاته مع تلك الناتجة عن محركات البحث على نفس الأطياف. يجادل المؤلفون بأن مستوى التغطية والتوافق هذا يعني أن التسلسل من الصفر، الذي كان يُعتبر لوقت طويل كطريقة احتياطية، يمكن أن يقف الآن إلى جوار بحث القواعد البيانية كخيار جدي، وفي بعض الحالات أفضل.
الصورة الكبيرة: نحو قراءة بروتينية كاملة وغير متحيزة
لغير المتخصص، الخلاصة أن DiNovo يجعل قراءة قطع البروتين بدقة أسهل بكثير دون أن تكون محدودة بما هو موجود بالفعل في قواعد المراجع. من خلال مضاعفة أو تضاعف كمية المعلومات المتسلسلة المدعومة جيداً وتوفير فحوصات خطأ مدمجة، يفتح هذا النهج الباب لاكتشاف بروتينات غير مألوفة، وتتبع تباينات طفيفة، واستكشاف خلطات معقدة حيث لا تزال مكوّنات كثيرة مجهولة. باختصار، من خلال إقران إنزيمات مرآوية مع التعلّم العميق وإحصاءات دقيقة، يساعد DiNovo في تحويل آثار الطيف المزعجة إلى صورة أوضح وأكثر موثوقية للبروتينات التي تقوم عليها الصحة والمرض.
الاستشهاد: Cao, Z., Peng, X., Zhang, D. et al. DiNovo enables high-coverage and high-confidence de novo peptide sequencing via mirror proteases and deep learning. Nat Commun 17, 2203 (2026). https://doi.org/10.1038/s41467-026-70224-6
الكلمات المفتاحية: علم البروتيوميات, التسلسل الببتيدي من الصفر, الطيف الكتلي, التعلّم العميق, البروتيازات المرآوية