Clear Sky Science · ar
تفعيل IRF3 في خلايا القلب يضعف الوظيفة التأكسدية للميتوكندريا عبر تثبيط PGC-1α ويدفع نحو فشل القلب
لماذا تهم القلوب المجهدة والخلايا المتعبة
غالباً ما يُوصَف فشل القلب بأن القلب «ينهار»، لكن القصة أعمق: هي أيضاً حكاية عن التهاب مزمن ومحطات طاقة منهكة داخل خلايا عضلة القلب. تطرح هذه الدراسة سؤالاً يبدو بسيطاً لكنه يحمل آثاراً كبيرة: هل هناك مفتاح جزيئي واحد داخل خلايا القلب يربط بين الالتهاب الضار وفشل إنتاج الطاقة—وإن وُجد، هل يمكن أن يغيّر تبديله مسار فشل القلب؟ عبر تتبع هذا الخيط، يكشف المؤلفون عن لاعب رئيسي ويُظهرون أن تعزيز برنامج الطاقة الداخلي للقلب بصورة معتدلة يمكن أن ينقذ جزئياً القلوب المتدهورة في الفئران.
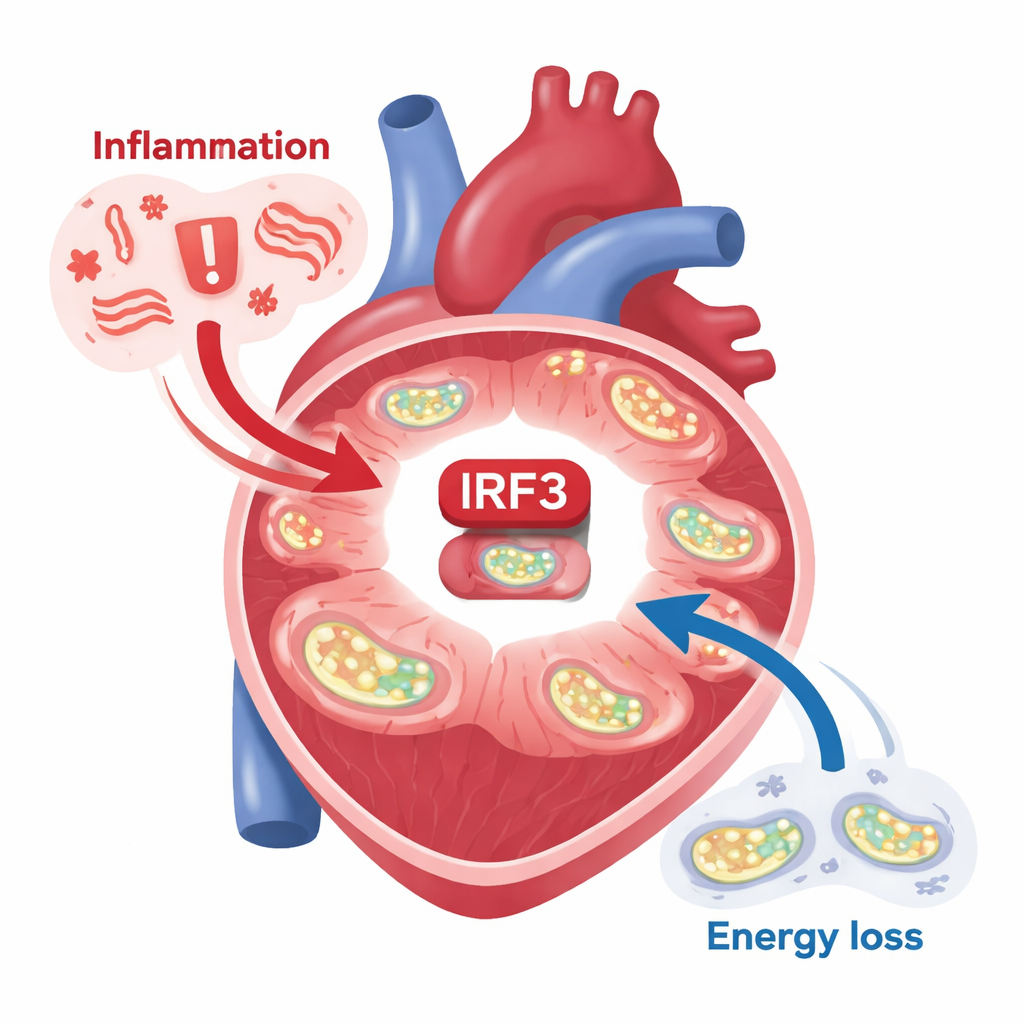
مفتاح جزيئي في قلوب بشرية مريضة
ركّز الباحثون على بروتين اسمه IRF3، المعروف بدوره في مساعدة الخلايا على الاستجابة للعدوى الفيروسية. فحصوا نسيجاً من أشخاص يعانون اعتلال عضلة القلب الإقفاري، وهو شكل شائع من فشل القلب ناجم عن نقص تدفّق الدم بعد النوبات القلبية. في هذه القلوب المتداعية، لم يكن IRF3 موجوداً فحسب—بل كان مفعلًا كيميائياً في مواقع محددة، وهو دليل على أنه يُشغّل برامج جينية. في الوقت نفسه، تدهور جهاز الميتوكندريا المسؤول عن تحويل الوقود إلى طاقة عبر الفسفرة التأكسدية بشكل واضح. ظهر نمط مشابه في نماذج الفئران لاحتشاء القلب: عندما جرى ربط شريان تاجي، تنبّه IRF3 في خلايا عضلة القلب بقوة، واضطرت الجينات التي تتحكم بها IRF3 إلى العمل. حتى شظايا من الحمض النووي الميتوكوندري—التي تُطلقها الميتوكوندريا التالفة وتعمل كإشارات «خطر» داخلية—كانت كافية لتفعيل IRF3 في خلايا القلب المعزولة.
إيقاف IRF3 يحمي القلب
لاختبار ما إذا كان نشاط IRF3 في خلايا عضلة القلب يفاقم المرض فعلاً، صمّم الفريق فئراناً يمكن إزالة IRF3 منها فقط في خلايا القلب، مع ترك خلايا المناعة والخلايا الداعمة الأخرى سليمة. بعد إحداث احتشاء قلبي، كانت وظيفة الضخ لدى هذه الفئران أفضل وكمية النّدبات أقل مقارنة بالفئران العادية، رغم تساوي الإصابة الأولية. في خلايا القلب المزروعة في الأطباق، أدى إسكات IRF3 إلى تهدئة الجينات الالتهابية من دون التأثير على بروتينات قريبة ذات صلة. مجتمعة، تشير هذه النتائج إلى أن IRF3 داخل خلية القلب ليس متفرجاً: بل يضخم الالتهاب والضرر البنيوي بعد الإقفار ويساهم في الانتقال نحو فشل القلب.
عندما يظل IRF3 في حالة «تشغيل»، ينهار نظام الوقود
قلب المؤلفون التجربة عكسياً: صنعوا فئراناً يمكن إجبار IRF3 في خلايا القلب على البقاء في حالة نشطة دائمة باستخدام حيلة جينية ذكية تُحاكي الفسفرة. حتى من دون محفز خارجي، طوّرت هذه الفئران بسرعة اختلالاً شديداً في وظائف القلب، ومستويات عالية من رسائل التهابية في الدم، وعلامات إصابة خلوية. كشفت دراسة معمقة لأنسجتها أن IRF3 النشط بصورة مزمنة يثبط منسق الطاقة الرئيسي المسمى PGC-1α. هذا الجزيء عادةً يعزّز صحة الميتوكوندريا والحرق الفعّال للدهون وتوازن طاقة الخلية. مع انخفاض PGC-1α، تقلدت عدة بروتينات ميتوكوندرية، ضعفَت سلسلة نقل الإلكترون، وتحولت خيارات الوقود في القلب: انخفضت الكارنيتين والمركبات المرتبطة باستهلاك الدهون، تعرّض استخدام الكيتونات للإضعاف، وتشوّه التعامل مع الجلوكوز. حتى نسبة NAD⁺ إلى NADH—مؤشر مهم لتوازن الأكسدة والاختزال الخلوي—انحرفت في الاتجاه الخاطئ.
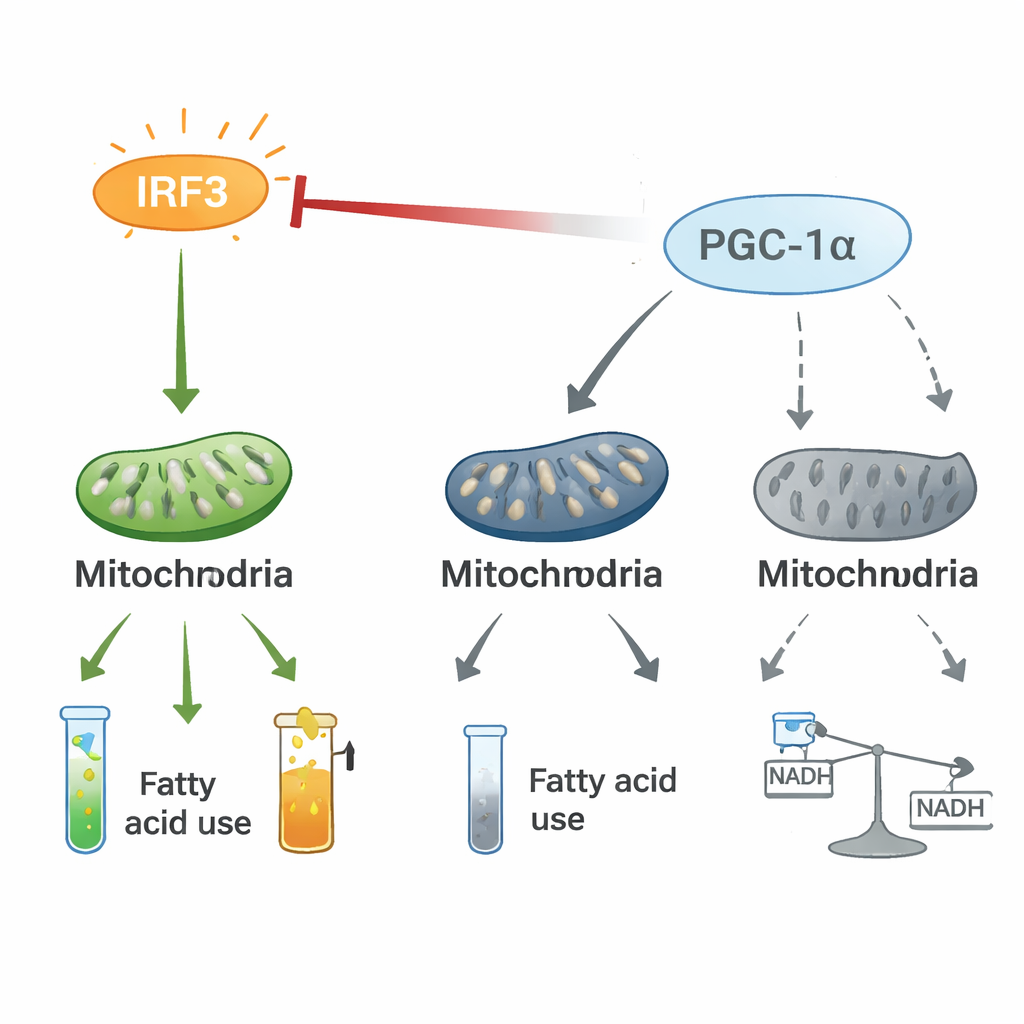
شد وجذب بين الالتهاب وضبط الطاقة
أوضحت تجارب آلية أن IRF3 وPGC-1α يشكلان محوراً تنظيمياً ثنائياً. في خلايا القلب، يرتبط IRF3 المُنشط جسدياً مع PGC-1α ويضعف قدرته على تشغيل جينات حرق الدهون. يؤدي خفض IRF3 إلى ارتفاع مستويات ونشاط PGC-1α، بينما يؤدي تعزيز PGC-1α إلى كتم الجينات الالتهابية التي يحفّزها IRF3 واستعادة علامات الميتوكوندريا، حتى تحت ظروف إجهاد مثل انخفاض الأكسجين أو السموم البكتيرية. أظهر تتبّع النظائر المستقرة أن تفعيل IRF3 يعيد توجيه الكربون من إنتاج الطاقة المعتاد عبر دورة حمض الستريك نحو مسارات بديلة مثل مسار فوسفات البنتوز، ويعطّل التدفق السلس للمتعاطيات الأيضية. هذا الشد والجذب بين مفتاح مؤيد للالتهاب (IRF3) ومساعد طاقة (PGC-1α) يبدو أنه يعيد تشكيل أيض القلب بطرق تُفضّل الالتهاب وفقدان الطاقة.
إعادة شحن معتدلة لبطاريات القلب
أخيراً، تساءل الفريق ما إذا كان دفع PGC-1α إلى الأعلى يمكن أن يعاكس أضرار IRF3. استخدموا ناقل علاج جيني موجه للقلب لرفع PGC-1α بشكل معتدل—لا مفرط—في نفس الفئران التي تملك IRF3 مفرط النشاط. حسّن هذا الرفع المتواضع وظيفة الضخ، وزاد البروتينات الميتوكوندرية، وعزّز الجينات المتعلقة بحرق الدهون واستقلاب NAD، وخفّض نشاط الجينات الالتهابية والليفية. في تجارب خلوية، أعاد التعبير المشترك عن PGC-1α مع IRF3 الناشط توازناً صحياً في نسبة NAD⁺/NADH وحوّل استخدام الوقود مرة أخرى نحو الدهون. للقارئ العام، يعني هذا أن إعادة شحن «نظام إدارة البطارية» في القلب بعناية يمكن أن تعوّض جزئياً الآثار الضارة لمفتاح التهابية خلوي زائد النشاط العالق في وضع «تشغيل».
ماذا يعني هذا لرعاية فشل القلب مستقبلاً
تضع هذه الدراسة IRF3 كرابطة مركزية بين الالتهاب وفشل الطاقة داخل خلايا عضلة القلب. بدلاً من التعامل مع الالتهاب والأيض كقضيتين منفصلتين في فشل القلب، تقترح الدراسة أنهما مرتبطتان عبر محور IRF3–PGC-1α. ورغم أن هذه النتائج مستمدة من فئران وخلايا، فإنها تفتح الاحتمال بأن العلاج المستقبلي قد يستهدف خفض نشاط IRF3 أو تعزيز PGC-1α ووظيفة الميتوكوندريا لإبطاء أو منع فشل القلب بعد النوبة القلبية. ببساطة، تهدئة نظام الإنذار الخلوي المفرط النشاط ودعم مصانع طاقة القلب قد يثبت أنه استراتيجية مشتركة قوية للحفاظ على قلوب أضعف تعمل بقوة لفترة أطول.
الاستشهاد: Kumari, M., Evangelakos, I., Deshpande, A. et al. Activation of IRF3 in cardiomyocytes impairs mitochondrial oxidative function through PGC-1α inhibition and drives heart failure. Nat Commun 17, 2051 (2026). https://doi.org/10.1038/s41467-026-69792-4
الكلمات المفتاحية: فشل القلب, الالتهاب, الميتوكندريا, خلايا القلب, PGC-1α