Clear Sky Science · ar
حالة مثيلة بروتين الفسفاتاز 2A تؤثر على اعتلالات α-سينوكلين في نماذج الفئران
لماذا هذا مهم لصحة الدماغ
مرض باركنسون والاضطرابات المرتبطة به تجرد الناس تدريجياً من الحركة والذاكرة والاستقلالية. مُذنِب رئيسي هو بروتين دماغي يُدعى ألفا-سينوكلين الذي يمكن أن يطيء، ويتكتّل، ويتلف الخلايا العصبية. تطرح هذه الدراسة سؤالاً مشجعاً: بدلاً من مهاجمة البروتين مباشرة، هل يمكننا ضبط آليات التنظيف الذاتية في الدماغ لمنع ألفا-سينوكلين من أن يصبح ساماً؟
حكاية بروتين لزج
في مرض باركنسون والخرف المصحوب بأجسام لوي، تتجمع خُصل من ألفا-سينوكلين الملتوية داخل الخلايا العصبية، مكوِّنة ما يُعرف بأجسام لوي التقليدية. وُسم كيميائي محدد على هذا البروتين، يضاف في موقع يُسمى سيرين 129، مرتبط بقوّة بأشكاله الأكثر ضرراً. عندما يكون هذا الوسم وفيراً، يميل ألفا-سينوكلين لتشكيل ألياف صلبة وتكتلات. يوازن الدماغ عادةً مثل هذه الوسوم باستخدام إنزيمات تضيفها وإنزيمات تزيلها. ونظراً لأن العديد من الإنزيمات يمكنها إضافة الوسم، فمن غير المرجح أن ينجح حظر إنزيم واحد فقط. لذلك ركز المؤلفون على عائلة الإنزيمات الرئيسية التي تزيل الوسم، المسماة بروتين فسفاتاز 2A أو PP2A، والتي تعمل كممحاة جزيئية لهذا التعديل الخطير.
ممحاة الدماغ ومفتاحاها الإثنان
لا يعمل PP2A بكامل قوته افتراضياً. تعتمد نشاطيته على وسم كيميائي صغير يُدعى المثيلة على أحد وحداته الفرعية. بروتينان آخران يتحكمان بهذا المفتاح: LCMT-1 يضيف الوسم ويحوّل PP2A إلى شكل أكثر نشاطاً وحماية، بينما PME-1 يزيله ويدفع PP2A نحو حالة أقل نشاطاً وأكثر ضرراً. أظهرت أعمال سابقة على أنسجة دماغية بشرية أنه في باركنسون وخرف أجسام لوي، يميل مستوى LCMT-1 إلى الانخفاض وPME-1 إلى الارتفاع، مما يترك PP2A مضعفاً. تختبر الدراسة الحالية مباشرة ما يحدث عندما تُدفع هذه المفاتيح عمداً في أي اتجاه في فئران حية تطور مشكلات ألفا-سينوكلين.
اختبار التوازن في أدمغة حية
استخدم الباحثون نموذجَي فئران مكمّلين. في أحدهما، تم تعديل الفئران وراثياً لإنتاج ألفا-سينوكلين البشري في جميع أنحاء الدماغ، فتتكوّن عندها تدريجياً تكتلات البروتين ومشكلات في الحركة والذاكرة مع التقدّم في السن. ثم عُدِّلت هذه الحيوانات أكثر لإنتاج كميات زائدة إما من PME-1 (مفتاح إيقاف PP2A) أو من LCMT-1 (مفتاح تشغيل PP2A) في خلايا قشرية أمامية. في النموذج الثاني، حقن الفريق ألياف ألفا-سينوكلين مُشكَّلة مسبقاً في المِقعَد القاعدي (السترياتيوم)، وهي منطقة دماغية عميقة متورطة في الحركة. تعمل هذه الألياف كَبـذور، فتجند ألفا-سينوكلين الطبيعي وتنشر المرض على مدى أشهر في فئران طبيعية أو مُعدَّلة إنزبيمياً. في كلا النموذجين، قاس العلماء تراكم البروتين، وصحة الخلايا العصبية، والتهاب الدماغ، والسلوك.
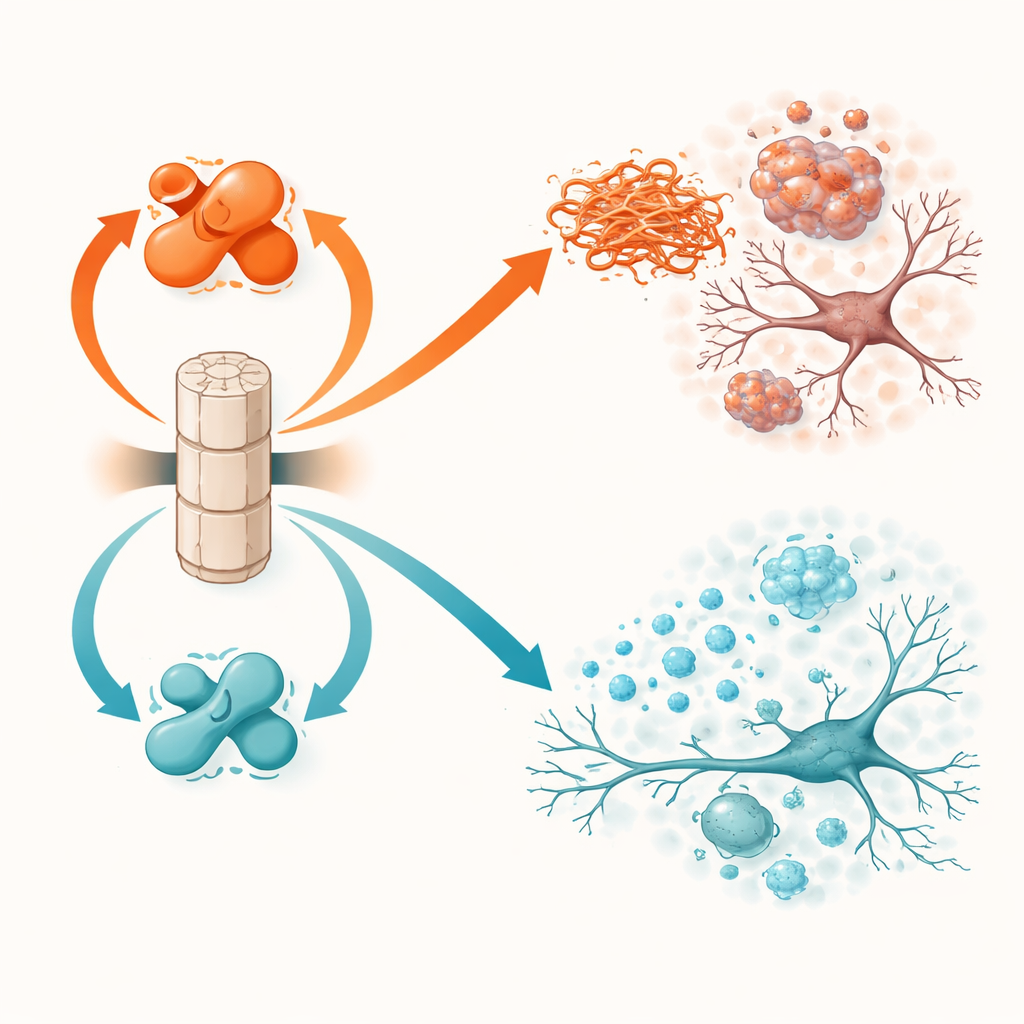
عندما تعطل الممحاة، ينتشر الضرر
الفئران التي أنتجت كميات زائدة من PME-1، وبالتالي كان لديها PP2A أقل نشاطاً، أدت حالتها إلى نتائج أسوأ. في الحيوانات المتحولة التي تعبر عن ألفا-سينوكلين، أدى تعزيز PME-1 إلى مزيد من الألفا-سينوكلين الموسوم والمتكتل في القشرة والحصين، فقدان أكبر لبنية الخلايا العصبية، إشارات نشاط عصبي أضعف، وتنشيط أقوى لخلايا المناعة في الدماغ. تُرجمت هذه التغيرات إلى أداء أسوأ في اختبارات الحركة ومهام التعلم والذاكرة. في نموذج الحقن بالألياف، سمح فرط التعبير عن PME-1 لتراكم تجمّعات ألفا-سينوكلين السامة وانتشارها بشكل أوسع، خصوصاً إلى الخلايا العصبية المنتجة للدوبامين في المادة السوداء، وهي منطقة رئيسية تتأثر في مرض باركنسون. أظهرت هذه الفئران فقداناً أشد لألياف الدوبامين، والتهاباً أقوى، وعيوباً أكبر في المهارات الحركية وبناء العش.
إعادة تشغيل الممحاة
كانت المناورة المعاكسة، إنتاج كميات زائدة من LCMT-1 للحفاظ على PP2A مثيلاً ونشطاً بقوة، لها تأثيرات وقائية عامة. في فئران ألفا-سينوكلين المتحولة، خفّضت LCMT-1 عبء البروتين الموسوم والمتكتل إلى مستويات قريبة من الطبيعي وحافظت على بنية ونشاط الخلايا العصبية. كانت علامات الالتهاب أقل، وأدى الأداء السلوكي للحيوانات إلى نتائج أقرب إلى الضوابط السليمة في اختبارات التوازن والذاكرة. في نموذج البذور بالألياف، حدّت LCMT-1 من كل من التراكم المحلي والانتشار طويل المدى لألفا-سينوكلين السام، وحمت خلايا الدوبامين من التحلل، وخفّضت تنشيط الخلايا البلاعمية الدقيقة، وقلّلت التدهور في التناسق الحركي وسلوك بناء العش. عبر التجارب، تُرجِمَ تحويل PP2A نحو حالته النشطة والمثيلة فوائد جزيئية إلى حماية وظيفية متسقة.
ماذا قد يعني هذا للعلاجات المستقبلية
للغير متخصصين، الدرس واضح: للدماغ ممحاة مدمجة يمكنها نزع وسم ضار عن ألفا-سينوكلين ومنعه من التحول إلى تكتلات خطيرة. عندما تضعف هذه الممحاة، يزداد الضرر والالتهاب والأعراض؛ وعندما تُقوّى، تُحاط الخلايا العصبية بالوقاية. توفر الدراسة دليلاً مباشراً في حيوانات حية على أن حالة مثيلة PP2A هي عنصر تحكم رئيسي في سمية ألفا-سينوكلين وتبعاتها. يشير هذا إلى استراتيجية علاجية جديدة: بدلاً من ملاحقة كل شكل ضار من البروتين، يمكن تصميم أدوية لتحفيز PP2A ومنظميه LCMT-1 وPME-1 نحو حالة أكثر وقاية. ستتطلب مثل هذه المقاربات اختبارات سلامة دقيقة، لكنها تحمل وعداً بإبطاء أو منع مرض باركنسون والحالات المرتبطة به عن طريق استعادة قدرة الدماغ الذاتية على ضبط ألفا-سينوكلين.
الاستشهاد: Maddila, S., Hassanzadeh, K., Liu, J. et al. Protein phosphatase 2A methylation state impacts α-synucleinopathy in mouse models. Cell Death Discov. 12, 172 (2026). https://doi.org/10.1038/s41420-026-03045-7
الكلمات المفتاحية: مرض باركنسون, ألفا-سينوكلين, بروتين فسفاتاز 2A, الضمور العصبي, التهاب الدماغ