Clear Sky Science · ar
تعزيز نشاط KLF15 في خلايا القلب العضلية: نهج جديد لمنع إعادة البرمجة المرضية والتليف عبر dCas9VPR الخالي من النشاط النوويزي
إعادة برمجة القلب المتدهور
يفرز فشل القلب ملايين الأشخاص، وغالبًا ما يتطور ببطء بعد سنوات من ارتفاع ضغط الدم أو أمراض الصمامات. في هذه الحالات، لا تزداد خلايا عضلة القلب حجمًا فحسب، بل تُشغِّل أيضًا برنامجًا وراثيًا "جنينيًا" وتمتلئ القلب بأنسجة ندبية. تستكشف هذه الدراسة طريقة جديدة لدفع آليات التحكم الجيني في القلب نحو حالة أكثر صحية—دون قطع الحمض النووي—من خلال رفع مستوى منظم وقائي يُدعى KLF15 بلطف داخل خلايا عضلة القلب.
عندما تفقد خلايا القلب هويتها
في القلب البالغ السليم، تحرق الخلايا القلبية الدهنية بكفاءة للحصول على الطاقة وتحافظ على نمط ثابت من نشاط الجينات. باستخدام تسلسل RNA أحادي الخلية في الفئران المعرَّضة لتحميل ضغط مزمن، رسم الباحثون خريطة التغيرات التي تطرأ على كل خلية قلبية أثناء انتقال القلب من وظيفة طبيعية إلى تضخّم ثم فشل. وجدوا أن عامل النسخ المسمى KLF15، الذي يحافظ عادة على توازن الأيض والنمو، أظهر أكبر تغير في النشاط في الخلايا المريضة. مع ازدياد الضغط، انخفضت مستويات KLF15 وضعفت قدرته على كبح الجينات الجنينية وتلك المرتبطة بالإجهاد. لوحظت تراجعات مماثلة في KLF15 في قلوب بشرية لمرضى اعتلال عضلة القلب التوسعي والتضخمي، مما يشير إلى أن هذا الاضطراب محفوظ عبر الأنواع.
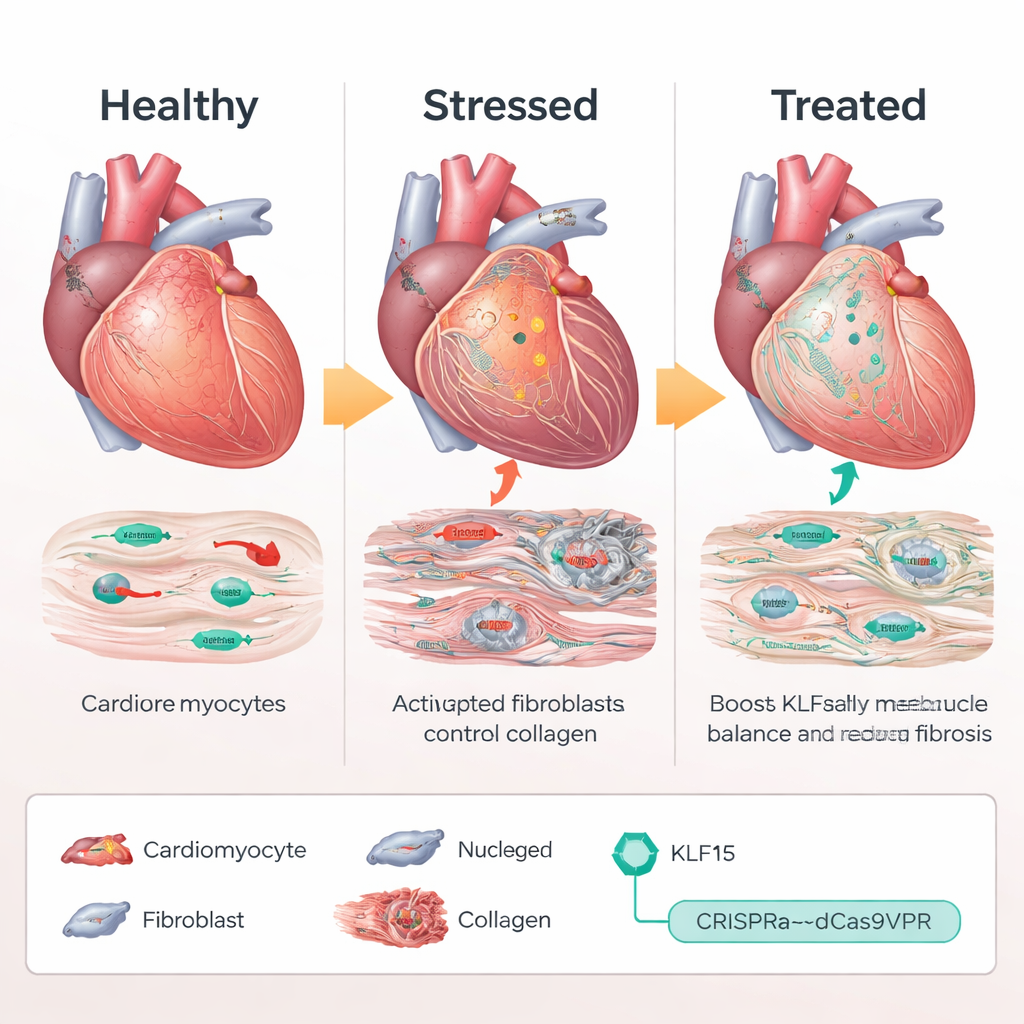
استخدام CRISPR كمفتاح ضبط صوت، لا كمقص
بدلاً من إضافة نسخة إضافية من جين KLF15 أو قطع الحمض النووي، استخدم الفريق نظام "تفعيل" قائم على CRISPR يُسمى dCas9VPR، يرتبط بالقرب من جين Klf15 الطبيعي ويعزز تعبيره الذاتي. في فئران مهندَسة للتعبير عن هذا المنشط CRISPR فقط في خلايا القلب العضلية، سلّم العلماء راوٍات الإرشاد عبر فيروس شبيه بالأدينو (AAV9) لاستهداف محفز Klf15. تحت حمل ضغط مزمن، حافظت الفئران التي تلقت دلائل تنشيط Klf15 على مستويات قريبة من الطبيعية لـ Klf15. بقيت خلايا عضلة القلب أصغر، تراجعت الوظيفة الضخية بدرجة أقل، وتحسنت النجاة مقارنةً بالحيوانات الضابطة. على المستوى الجزيئي، خفتت جينات الإجهاد والجنينية، بينما تعافت جينات الأيض ومعالجة الكالسيوم، مما يشير إلى أن البرنامج النسخي المرضي قد عُدّل إلى حد كبير.
إخماد تكون الندب عبر تواصل الخلية مع الخلية
لا يقتصر فشل القلب على الخلايا العضلية المريضة فحسب، بل يساهم أيضًا الأرومات الليفية، وهي خلايا داعمة تنتج الكولاجين وتشكل نسيجًا ندبيًا جامدًا. أظهرت تحليلات أحادية الخلية وتصوير الأنسجة أن استعادة Klf15 في خلايا القلب العضلية قللت من تنشيط الأرومات الليفية ومن التليف العام، رغم أن العلاج الجيني لم يستهدف الأرومات الليفية مباشرة. تتبَّع الفريق هذا الأثر إلى بروتين مُفرَز يدعى AZGP1. عندما ارتفع Klf15 في خلايا القلب العضلية، زاد إنتاج وإفراز AZGP1. في قلوب الفئران وأنسجة قلب بشرية مشتقة من الخلايا الجذعية، خفَّضت مستويات AZGP1 المنخفضة مسار TGF-β / SMAD في الأرومات الليفية—وهو محرك رئيسي للتندُّب—مما خفض مؤشرات مثل α-SMA وPOSTN. ومن المهم أن فرط تعبير AZGP1 في خلايا القلب وحده لم يُعدّل هوية الخلايا العضلية، مما يبيّن أن KLF15 يحمي خلايا القلب مباشرة ويستخدم AZGP1 كرسول للحد من نشاط الأرومات الليفية.
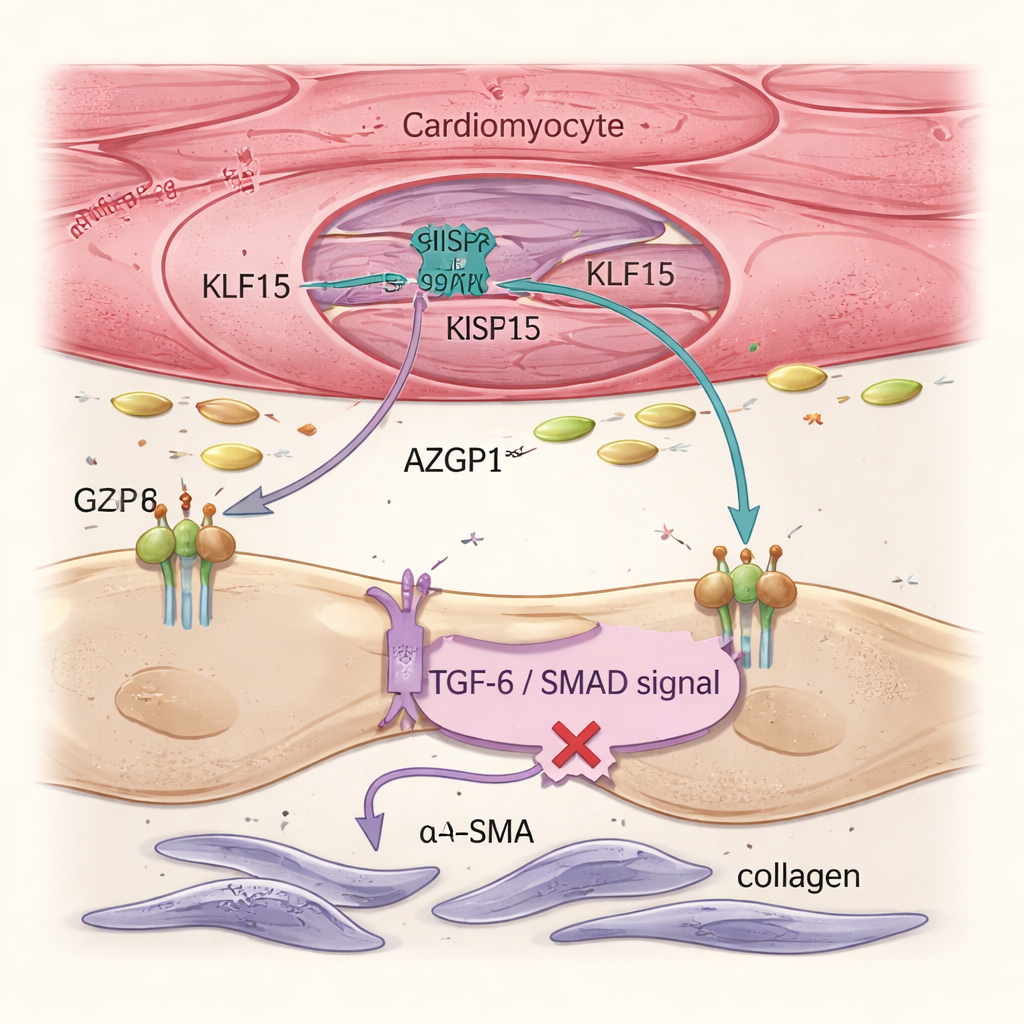
نماذج الأنسجة البشرية تؤكد الدائرة الوقائية
لاختبار ما إذا كانت هذه الآليات تنطبق على الخلايا البشرية، استخدم الباحثون خلايا قلبية مشتقة من الخلايا الجذعية المحفزة ومزروعة في أنسجة قلب هندسية ثلاثية الأبعاد. عند تعرّضها لحِمْل ميكانيكي يحاكي ارتفاع ضغط الدم، فقدت هذه الأنسجة KLF15، وأشعلت جينات الإجهاد والجنينية، وتجمدةّت، وضعفت تقلصاتها—مستنسخةً سمات المرض. منعت استعادة KLF15 المدفوعة بـ CRISPRa هذا التدهور، محافظةً على توليد القوة، ومحوِّلةً التعبير الجيني نحو أيض ونُظم بنيوية أكثر نضجًا. أظهرت تجارب مفصلة أن TGF-β1، الإشارة المعروفة المحفزة للتليف، تقلل KLF15 في الخلايا القلبية البشرية عبر مسار SMAD2/3، ما يساعد على تفسير كيف يؤدي الإجهاد المزمن إلى إعادة تشكيل ضارة. أخيرًا، صمم الفريق نظام CRISPRa مُصغَّر قائم على نسخة أصغر من Cas9 تتسع في ناقل AAV9 واحد ويُشغّل بواسطة محفز خاص بخلايا القلب العضلية. في شرائح دقيقة مقطوعة بدقة من نسيج قلب بشري متدهور، رفع هذا الناقل بنجاح مستويات KLF15 وحسّن الأداء الانقباضي على مدار أيام في الثقافة.
مخطط لعلاج جيني ألطف
للقارئ غير المتخصص، الرسالة الأساسية هي أن هذا العمل يُظهر كيف يمكن للرفع الحذر لمنظم وقائي واحد داخل خلايا عضلة القلب أن يثبت هويتها ويرسل إشارات تقلل التندُّب. باستخدام منشط مستند إلى CRISPR لا يقطع الحمض النووي، تُعدّل الطريقة جينات القلب الطبيعية بدقة بدلًا من إدخال جين اصطناعي. تحدد الدراسة مسارًا TGF-β → KLF15 → AZGP1 يربط الإجهاد الميكانيكي بإعادة التشكّل الضارة وتُظهِر، في الفئران ونماذج الخلايا البشرية وشرائح نسيج القلب البشري، أن استعادة KLF15 يمكن أن تقاطع هذه السلسلة من الأحداث. وعلى الرغم من كونها لا تزال في مرحلة قبل السريرية، يقدم نظام CRISPRa المصغَّر والموجَّه إلى خلايا القلب هنا خارطة طريق محتملة لعلاج أشكال فشل القلب الشائعة وغير الوراثية عن طريق إعادة برمجة نشاط الجينات بدلًا من إعادة كتابة الجينوم.
الاستشهاد: Schoger, E., Kim, R., Bleckwedel, F. et al. Enhancing KLF15 activity in cardiomyocytes: a novel approach to prevent pathological reprogramming and fibrosis via nuclease-deficient dCas9VPR. Sig Transduct Target Ther 11, 76 (2026). https://doi.org/10.1038/s41392-026-02593-9
الكلمات المفتاحية: فشل القلب, KLF15, تفعيل CRISPR, تليف قلبي, AZGP1